
Abstract
Adult myocardium has the capacity for repair and regeneration, which is derived from cardiac stem cells (CSCs). In this study, we assessed the migration and changes in numbers of Sca-1-positive CSCs after myocardial infarction (MI) in vivo and in vitro. In this study, we showed that in a rat MI model the CSCs emerged around the vessels near the peri-infarct zone and in the epicardium of the infarcted area. Four weeks after infarction, no differences in the expression of connexin 43 (Cx43) were observed in the peri-infarct and infarct zones. In vitro, we mimicked tissue ischemia and hypoxia by using a culture environment of 5 % O2 and a wound healing assay to monitor the migration of CSCs. In conclusion, under hypoxic conditions, the CSCs, conveyed by blood vessels, migrated from the niche to the infarct zone for repairing the damaged myocytes. The number of endogenous migrating CSCs was proportionate to the repair time after infarction, rather than the degree of infarction. Four weeks after MI, the expression of Cx43 was not altered in migratory CSCs, namely no enhanced gap-junctional communication with cardiomyocytes was seen in the CSCs. Further studies are necessary to delineate the molecular mechanisms that drive the migration of CSCs after MI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The adult myocardium has the capability of repair and regeneration, a capacity that is derived from cardiac stem cells (CSCs) [1]. CSCs are self-renewing and clonal proliferation of stem cells was first described in 2002 in CSCs from mouse heart [2]. CSCs have the potential for pluripotent differentiation into a variety cells such as cardiomyocytes, endothelial cells, and vascular smooth muscle cells [3–5].
Torella et al. [6] discovered that CSCs have the ability to maintain a steady state of the myocardial cells involved in heart disease-induced remodeling, which may be the basis of CSC-derived myocardial repair. Thereafter, the repair potential of CSCs has been discovered not only in myocardial ischemia but also in myocardial infarction (MI). Messina et al. injected CSCs into a mice MI model, and then observed their differentiation into myocardial cells, and vascular endothelial and smooth muscle cells, and the subsequent repair of the necrotic myocardium and improvement of ventricular function [7].
In subsequent studies, CSCs have been purported to be important cardiac repair cells. Rossini et al. demonstrated the differentiation abilities of these CSCs and that of the conventionally used bone marrow-derived MSCs [8]. They showed that despite the fact that CSCs were less able to acquire osteogenic and adipogenic phenotypes, they expressed cardiovascular markers more efficiently. Moreover, CSCs showed longer survival when transplanted into the infarcted heart and a better ability to differentiate into cardiomyocytes than bone marrow-derived MSCs [9].
Studies also showed that CSCs have the effect of maintain myocardial cell homeostasis and participate in cardiac remodeling [10, 11]. The mechanism by which a limited number of CSCs migrate and reach the necrotic area for regeneration after MI is unknown and few studies have addressed this. Kuang et al. [12] showed that MI of the rat heart led to an increased expression of stem cell factor (SCF), which mediated the migration of CSCs via stimulation of c-kit and activation of the p38 mitogen-activated protein kinase (MAPK). The effect of an ischemic myocardial event on the cellular metabolism and homeostasis of CSCs is also unknown. In this study, we monitored the changes in the migration and proliferation of Sca-1-positive CSCs after MI and characterized the migratory mechanism in vivo and in vitro. We compared the position and numbers of CSCs in vivo before and after MI by pathological examination. We also mimicked tissue ischemia and hypoxia in vitro using a wound healing assay.
MATERIALS AND METHODS
Animals and Experimental Model
Thirty-two adult male Wistar rats, weighing 200–250 g, were obtained from the Laboratory Animal Science Department, the Second Affiliated Hospital of Harbin Medical University, Heilongjiang, China. All experimental animal procedures were approved by the Local Ethical Committee of Harbin Medical University Animal Care and Use. Animals were housed in controlled environmental conditions in a pathogen-free facility, exposed to 12-h light/dark cycles, and had access to a standard diet and tap water. The rats were randomly placed in one of four groups; rats without operation (Baseline group), rats that were allowed to survive 1 week after a coronary artery ligation operation (1w), rats that were allowed to survive 2 weeks after a coronary artery ligation operation (2w), and rats that were allowed to survive 4 weeks after a coronary artery ligation operation (4w).
The acute myocardial infarction (AMI) model was established by left coronary artery ligation according to the protocols described in a previous report [13]. Rats were anesthetized with an intraperitoneal injection of 10 % chloralhydrate (4 ml/kg, i.p.). After endotracheal intubation and initiation of ventilation (room air, rate 80 cycles/min, tidal volume 1 ml/100 g of body weight; Harvard Apparatus Rodent Ventilator, model B14963) under sterile conditions, left intercostal thoracotomy was performed and the third and fourth intercostal ribs were separated with a small retractor to expose the heart. The pericardium was opened and the left anterior descending coronary artery (LAD) was permanently ligated with a 6–0 polyester suture 1 mm from tip of the normally positioned left auricle. Successful ligation of the LAD was verified by myocardial blanching and abnormal movement of the anterior wall. Then, the chest cavity was closed and gas was pumped gas in the chest.
Isolation and Culture of CSCs from Adult Mouse Heart
CSCs were isolated from the hearts of BALB/c mice (18–25 g) by a method described previously, with a minor modification [1]. Briefly, the mouse was injected with heparin (5,000 IU/kg, i.p.) 20 min prior to the experimental protocol, and then was sacrificed by cervical dislocation. The heart was excised and the aorta was cannulated rapidly. The cannulated heart was mounted on a Langendorff perfusion apparatus with constant flow and the perfusion pressure was monitored. The heart was first perfused with calcium-free Tyrode’s solution for 10 min to remove the blood and then digested with 0.5 mg/ml collagenase (Sigma, Poole, UK) and 0.5 mg/ml trypsin (GIBCO) at 37 °C for 30 min. After the enzymatic digestion was terminated, the heart tissue was cut into pieces, suspended in with Dulbecco’s modified Eagle medium (DMEM)/F12 (HyClone, Logan, UT) containing 10 % fetal bovine serum (HyClone) and the cell suspension was filtered with a Steriflip (Millipore). Then, the cells were incubated with a fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse Sca-1 antibody (BD Pharmingen, San Diego, CA) and separated by using anti-FITC microbeads for magnetic cell sorting (MACS). Small round cells, containing most of the Sca-1-positive cell population, were separated. These Sca-1-positive cells were cultured for 3–5 days with DMEM/F12 (HyClone) containing fetal bovine serum (HyClone), 10 ng/ml basic fibroblast growth factor (bFGF) (Peprotech, Rocky Hill, NJ), 10 ng/ml insulin-like growth factor (IGF) (Peprotech), 10 ng/ml epidermal growth factor (EGF) (Peprotech) and 10 ng/ml leukemia-inhibitory factor (LIF) (Sigma) at 37 °C. The cells were used for subsequent experiments after a recovery period.
Echocardiography
Transthoracic echocardiography (VIVIDT 7, GE; equipped with a 10-MHz phased-array transducer) was performed to assess the changes in cardiac function in the four groups of rats. The hearts (n = 8 per group) were imaged in 2D and in the M-mode, and recordings were obtained from the parasternal long-axis view at the papillary muscle level (Table 1).
Immunohistochemistry
Rats belonging to the 1w, 2w, and 4w groups were sacrificed in the first, second, and fourth weeks after ligation of the LAD. The heart tissue below the occlusion site was collected and embedded in optimal cutting temperature medium (OCT; Tissue-Tek, France) and 8-μm frozen sections (n = 8 per group) were prepared. To detect the expression of Sca1 in the heart tissues, a rabbit antibody (1:400, ab95439; Abcom) against Sca1 was used. Endogenous peroxidase was blocked by treating the sections with 0.3 % H2O2 for 20 min. A biotin-conjugated anti-rabbit immunoglobulin (1:600; Dako) secondary antibody was used for immunostaining.
Immunofluorescence
For immunofluorescence, the heart tissue below the occlusion site of the sacrificed rats was fixed in 4 % formalin and 5-μm paraffin sections (n = 8 per group) were prepared. The sections were allowed to thaw at room temperature for at least 2 h and then fixed in cold acetone for 2 min. Primary anti-connexin 43 (Cx43) (1:200) antibodies were used. After 3- to 5-min washings, sections were incubated with a mixture of Rhodamine (TRITC) goat anti-rabbit antibody (1:50) for 1 h in 1 % blocking buffer. After incubation for 15 min in DAPI stain, the sections were washed and examined under the microscope.
Immunofluorescence detection was performed with a laser scanning confocal microscope (Leica, Inc). The CSCs that were positive for Cx43 were counted using the Image-Pro-Plus software (version 6.0; National Institute of Health) in at least five high-power fields (HPF, ×400) in the infarct and peri-infarct regions, which were randomly selected and counted in at least three sections from each animal (n = 8 per group).
Masson’s Trichrome and Hematoxylin and Eosin Staining
Masson’s trichrome stain was used to quantify the extent of fibrosis and infarct size in the left ventricle (LV). The fibrotic and total areas of the LV on each image were measured using the Image-Pro-Plus software, and the percentage fibrotic area was calculated as (fibrotic area/total LV area) × 100 %. For routine histopathology, heart sections were stained with hematoxylin and eosin (H&E). At least five thin sections from each heart were stained.
Flow Cytometry Analysis
To determine the phenotype of the Sca-1-positive population of CSCs by flow cytometry, single-labeling methods were employed for defining different subpopulations. The cells, at passage 3 from the MACS isolation, were washed with cold phosphate-buffered saline (PBS) containing 1 % bovine serum albumin, centrifuged, and resuspended in cold PBS. The cells were then stained with rat anti-mouse Sca-1 (BD, USA). Cells were incubated with the antibody for 30 min at 4 °C and washed three times with cold PBS. Samples were analyzed using a FACScan system (BD Biosciences).
In Vitro Migration Assay and Hypoxia
Cell migration was assessed using wound-healing assays. Cultures of Sca-1-positive CSCs in 6-well culture plates were scratched with 10-μl pipette tips to create fixed-width linear “wounds” in the cell monolayers. The hypoxia was achieved by switching a perfusing solution from a bath solution equilibrated with 5 % O2, 5 % CO2, and balance with N2 (normoxic). The cells were culture under the conditions of 95 % O2 as control group. Cell migration was monitored by capturing the images of linear wound closure using a 2× bright field objective in an Olympus 1 × 70 inverted microscope coupled to a Sony camera. Images were taken immediately after wounding and at defined time intervals thereafter, until complete closure was observed in the control cultures.
Statistics Analysis
Values for all measurements are presented as mean ± SD. Data for two different groups were compared using the least significant difference (LSD) test. Comparisons of means of multiple groups were performed by analysis of variance (ANOVA). A value of p < 0.05 was considered statistically significant. Statistical analysis was performed with the SPSS (version 18.0; Chicago, IL) software package.
RESULTS
The AMI Model
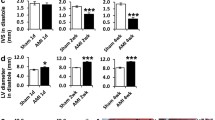
Echocardiography imaging was performed in the four groups of rats (Fig. 1). At various time points, the groups that had the operation had significant differences compared to the baseline group (Table 1). The left ventricular ejection fraction (LVEF) and the left ventricular fractional shortening (LVFS) were significantly reduced in groups that had the operation compared to the baseline group (Fig. 1).

Echocardiographic assessment of cardiac function. Representative M-mode echocardiograms are shown at baseline (normal rats) and in the 1w, 2w, and 4w groups. In the groups that had surgery, 1, 2 and 4 weeks after ligation of the LAD, the left ventricular ejection fraction (LVEF) declined to 45.49 ± 4.05 %, 46.51 ± 8.58 %, and 52.88 ± 0.71 %, respectively, compared with the baseline (83.45 ± 2.99 %, p < 0.01). The left ventricular fractional shortening (LVFS) declined to 19.49 ± 1.94 %, 20.20 ± 4.54 %, and 23.47 ± 0.48 %, 1, 2, and 4 weeks after AMI, respectively, compared with the baseline (46.66 ± 3.57 %, p < 0.01). **p ≤ 0.001, compared to the baseline group; n = 8 for each group.
Furthermore, Masson’s trichrome staining showed transmural infarction in all groups (Fig. 2a). There was enhanced extracellular matrix accumulation and fibrosis in the groups with surgery compared to the baseline group. As expected, the LV fibrotic area was markedly increased in groups 1, 2 and 4w after ligation of the LAD, compared to the baseline group (p < 0.05, p < 0.01, and p < 0.01, respectively).

Masson's Trichrome staining of the left ventricle (LV) (a), the infarct zone (b(a)) and the peri-infarct zone (b(b)). The images of the LV (×40) captured after Masson's trichrome staining show a significantly greater fibrotic area in the 1w (a(a)), 2w (a(b)) and 4w (a(c)) groups than in the baseline group (a(d)). The fibrotic areas in the LV were 14.3 ± 7.77 % (a(a)), 26.7 ± 10.4 % (a(b)), and 23.3 ± 10.4 % (a(c)) in the 1w, 2w, and 4w groups, respectively. **p < 0.01, compared to the baseline group (a(d)). Scale bars represent 1.0 mm. Images (×40) of the infarct and peri-infarct zone (b) captured after Masson's trichrome staining showed significantly higher myocardial collagen concentration in the infarct zone (b(a), 24.8 ± 7.08 %) as compared with the peri-infarct zone (b(b)), 5.16 ± 1.71 %, p < 0.01). **p < 0.01, compared to the peri-infarct zone (b(b)). Scale bars represent 1.0 mm.
The development of inflammation, with the infiltration of numerous leukocytes, was observed by H&E staining in the infarcted hearts (Fig. 3a). Increased inflammatory cell infiltration was observed in the groups with surgery, compared to the baseline group (Fig. 3a). These results suggested that the AMI model was established successfully and fibrosis and inflammation occured as expected in the infarcted hearts.

Hemotoxylin- and eosin-stained rat myocardium after LAD ligation. Increased inflammatory cell infiltration was observed in 1w (a(a, a′)), 2w (a(b, b′)) and 4w (a(c, c′)) groups, compared with that in the baseline group (a(d, d′)). Increased inflammatory cell infiltration was observed in the infarct zone (b(a)), compared with that in the peri-infarct zone (b(b)). Scale bars represent 200 μm.
We also observed some additional changes. [1] The results of Masson's trichrome staining showed significantly larger fibrotic areas in obesity infarct zone than in the peri-infarct zone (Fig. 2a). [2] The muscle fiber appeared to be in an non-structured state and the cardiocytes exhibited nuclear pyrosis, nuclear fragmentation, and even disappeared in the infarct zone. Part of the myocardial tissue appeared to have focal and patchy necrosis, obvious inflammatory cell infiltration, and the cardiac fibrous structure was chaotic in the infarct zone (Fig. 3b(a)). [3] Normal heart muscle tissue and infarcted myocardial tissue existed alternately in the peri-infarct zone. In the normal heart muscle, every muscle fiber had horizontal stripes of tissue, as well as intercalated discs of myocardium, with an evident nucleus (Fig. 3b(b)).
Spatiotemporal Characterization of CSCs After AMI
To investigate the changes in endogenous CSCs after AMI in the cardiac tissue of the rats, the expression of Sca1, a CSC-specific marker, was detected in labeled cells using immunohistochemical staining. We observed a significantly higher increase in the number of CSCs in the vessels near the peri-infarct zone than in normal cardiac tissue.
Furthermore, CSCs migrated to the epicardium of the infarcted area after AMI. The increase in migration was 10-, 40- and 160-fold in the 1w, 2w, and 4w groups, respectively, compared to that in the baseline group (p < 0.001, Fig. 4b–e). It is of particular interest that the higher numbers of CSCs that migrated were also strongly associated with onset time of AMI. Because only a few blood vessels exist in the infarcted area, the results suggest that the migration of the CSCs into the infarcted area is not mediated by or dependent on blood vessels.

Immunohistochemical staining for Sca-1-positive CSCs. Sca-1-positive CSCs emerged around the vessels near the peri-infarct zone (a, arrows) and in the epicardium of the infarcted area, 1 week (b), 2 weeks (c), and 4 weeks (d) after infarction, or in normal cardiac tissue (e). The number of CSCs has increased around the vessels near the peri-infarct zone (6.83 ± 1.34, a) as compared with normal cardiac tissue (e, p < 0.01). The CSCs emerged in the epicardium of the infarcted area (5.33 ± 1.50, 20.6 ± 4.45, 78.1 ± 7.9) in the 1w, 2w, 4w groups, respectively) after AMI. Note that the number of CSCs that emerged in the epicardium of the infarcted area is largest in the 4w group.
Cx43 Expression in the Heart After AMI
To determine the relationship between the endogenous migratory CSCs and cardiomyocytes, the expression Cx43 in rat hearts after AMI was evaluated using immunofluorescence. Cx43 localized at intercalated disks of myocytes in the normal heart (Fig. 5a,a′). However, in the peri-infarct zone the expression of Cx43 are different in size and shape and disordered distributing (Fig. 5b,b′). Meanwhile, the number of Cx43 decreased remarkably in the infarct zone as compared with nomal heart tissue (Fig. 5c,c′). However, we found no significant differences in the expression of Cx43 in the hearts of the 1w, 2w, and 4w group rats (data not shown).

Immunostaining analysis of the expression of connexin 43 (Cx43) in rat heart tissues. Representative confocal micrograph images of Cx43 (magenta) in the normal zone (a, a′), the peri-infarct zone (b, b′), the infarct zone (c, c′), and the nuclei (DAPI, blue). Images shown in a′, b′, and c′ are at a higher magnification than that used for a, b, and c panels.
Isolation of Sca-1-Positive CSCs and the Effect of Hypoxia on the Motility of Sca-1-Positive CSCs
In view of our previous results that showed that the numbers of CSCs were increased significantly in the epicardium of the infarcted area and around the vessels near the peri-infarct zone (Fig. 4), we surmised that ischemia and hypoxia may be involved in driving the migration of endogenous CSCs. To further investigate the factors that induce the migration of endogenous CSCs after AMI, a wound healing assay was performed. First, we isolated Sca-1-positive CSCs from rat cardiac tissue using MACS. After isolation, the Sca-1-positive CSCs were cultured in conditioned medium. After the third passage, the cells were harvested and the proportion of Sca-1-positive CSCs was estimated by flow cytometry. The proportion of Sca-1-positive cells reached 83.5 ± 1.8 % (Fig. 6a).

FACS Analysis of Sca-1-positive cells (a). Sca-1-positive cells constitued 83.5 ± 1.8 % of the purified myocardial small cells. Effect of hypoxia on wound healing assay (b): 95 % O2 condition 0 h (a), 5 % O2 condition at 0 h (b), 95 % O2 condition at 24 h (c), and 5 % O2 condition at 0 h (d). Images were taken at the end points and compared to the 0 h samples to measure wound healing.
Second, a wound healing assay was performed to assess the motility of CSCs in an environment of 5 % or 95 % O2. The status of the cells s at 0 h in each group was regarded as the baseline for that group. We found that in the 5 % O2 condition, the Sca-1-positive CSCs migrated into the scratch (88.5 ± 3.02 % vs. baseline, p < 0.01; Fig. 6b). However, under the 95 % O2 condition there was no significant migration compared with the baseline (48.67 ± 5.09 % vs. baseline, p < 0.01; Fig. 6b). These observations indicate that a hypoxic condition was efficient in promoting the migration of Sca-1-positive CSCs.
DISCUSSION
The goal of this study was to investigate the migration and changes in the numbers of Sca-1-positive CSCs after myocardical infarction. We found that the numbers of Sca-1-positive CSCs increased significantly around the vessels near the peri-infarct zone and in the epicardium of the infarcted area after MI. This change in the numbers of CSCs was time-dependent and correlated with the infarction time. We also found that the migration of endogenous CSCs after AMI was likely not associated with the gap junction protein Cx43. Finally, hypoxia promoted Sca-1-positive CSC migration efficiently. Overall, our results showed that after MI, and induced by hypoxic conditions, endogenous CSCs migrate into the peri-infarct zone through blood vessels, and into the epicardium of the infarcted area independent of the blood vessels and the gap junction protein Cx43, enabling the repair of damaged tissues.
In the past, the heart was viewed as an organ composed of terminally differentiated cardiomyocytes and incapable of regeneration. Recent studies challenge these notions regarding cardiac repair/regeneration and suggest that the heart is capable of limited regeneration through the activation and recruitment of a stem/progenitor cell population that is resident in the adult heart. Many studies have described a CSC or progenitor cell population that may participate in limited myocardial regeneration in response to an injury. Bearzi et al. [14] discovered endogenous CSCs within structural and functional units known as niches. The cardiac niches are expected to control the physiological turnover of myocardial cells and the growth, migration, and commitment of primitive cells leaving niches to replace old dying cells in the myocardium. However, the mechanisms that mediate the migration of these CSCs into the injured myocardium after MI are not yet known. Urbanek et al. [15] found that the CSC pool is acutely enhanced after infarction. In this study, we monitored the changes in the numbers of endogenous CSCs after AMI in the cardiac tissue of rats. We found that CSCs crossed fibrotic regions in the infarct zone and reached the epicardium of the infarcted area. In addition, the number of endogenous CSCs in the epicardium of the infarcted area was markedly increased in the 4w group, followed by the increases in the 2w, and then the 1w group, compared to that in the baseline group. Endogenous CSCs also existed around the blood vessels in the peri-infarct zone, but these CSCs were fewer in number than those in the epicardium. Based on these data, we conclude that the number of migrating endogenous CSCs increases proportionate to the repair time after infarction, rather than the degree of infarction.
Fibrosis was markedly increased in the 2w and 4w groups compared to that in the 1w group, whereas fibrosis was lower in the 4w group compared to that in the 2w group. This prompts us to ask how the CSCs reached the epicardium of the infarcted area across the fibrosis. From the results of the Massons’ trichrome staining and immunohistochemical analysis, we surmise that after infarction the repair mechanisms involved in promoting the migration of endogenous CSCs to the injured regions are initiated. The process of migration of CSCs is likely to proceed in two steps. First, the endogenous CSCs arrive at the peri-infarct zone from the niche through the blood vessels. Then, CSCs migrate to the epicardium of the infarcted area from the peri-infarct zone around the vessel. The second step is perhaps driven by some cellular factors, since the number of vessels in the infarcted area is too few to convey the CSCs. Therefore, we surmise that the migration of the CSCs happens via two mechanisms, the “high-speed channel” and the “low-speed channel.” The blood vessels act like a “high-speed channel,” which allow migration of the CSCs to the peri-infarcted area from the niche over farther distances. Then a “low-speed channel” comes into play, and some as yet unknown cellular factors promote the migration of CSCs to the epicardium of the infarcted area. The “high-speed channel” can shorten the time of migration and facilitate quick repair; the “low-speed channel” can mobilize the CSCs to migrate into the infarcted area under a harsh environment, characterized by a paucity of vessels. After MI, the internal distribution of cells in cardiac tissue is also changed. The CSCs migrate from the niche to the infarct zone for repairing the damaged myocytes. When myocytes are severely damaged, the CSCs are mobilized and migrated into the infarct zone. The longer the repair time after infarction, of the larger the numbers of CSCs that are mobilized into the infarct zone to repair myocardium. Therefore,the study of repair mechanisms of the CSCs has important clinical significance in the diagnosis and treatment of heart disease, and further research into the molecules involved in these mechanisms is needed.
The CSC pool is enhanced acutely after infarction [15], and ischemia and hypoxia induce the injury and necrosis of myocardium in MI. We hypothesized that ischemia and hypoxia may impact endogenous CSC migration. We tried to mimic tissue ischemia and hypoxia using an in vitro wound healing assay performed under hypoxic conditions. Hypoxia efficiently induced the migration of Sca-1-positive CSCs. Further research is needed to clarify the mechanism by which ischemia and hypoxia induce endogenous CSCs migration.
To determine the relationship between CSCs and cardiomyocytes, we monitored the expression of Cx43, a cardiomyocyte-specific marker, using immunofluorescence. We found that the expression of Cx43 decreased remarkably in the infarct zone, compared with its expression in normal heart tissue, but there were no differences in the expression of Cx43 between the 1w, 2w and 4w groups of rats. These data indicate that 4 weeks after infarction, the migratory CSCs had not differentiated into and connected with cardiomyocytes, although they had migrated into the injured region. It is likely that CSCs have the capacity to repair and regenerate via paracrine effects 4 weeks after infarction.
Change history
12 August 2020
After publication of our article [1], we became aware that there were errors in Fig. 1, namely that the Echocardiographic assessment of 1w, 2w, and 4w groups was incorrectly presented.
Abbreviations
- CSC:
-
cardiac stem cell
- MI:
-
myocardial infarction
- AMI:
-
acute myocardial infarction
- LAD:
-
left anterior descending coronary artery
- MACS:
-
magnetic cell sorting
- LV:
-
left ventricle
- LVEF:
-
left ventricular ejection fraction
- LVFS:
-
left ventricular fractional shortening
- Cx43:
-
connexin 43
References
Epstein, J.A., and H. Franklin. 2010. Epstein Lecture. Cardiac development and implications for heart disease. N Engl J Med 363: 1638–1647.
Hierlihy, A.M., P. Seale, C.G. Lobe, M.A. Rudnicki, and L.A. Megeney. 2002. The post-natal heart contains a myocardial stem cell population. FEBS Lett 530: 239–243.
Linke, A., Muller, P., Nurzynska, D., Casarsa, C., Torella, D., Nascimbene, A., Castaldo, C., Cascapera, S., Bohm, M., Quaini, F., and other 5 authors. 2005. Stem cells in the dog heart are self-renewing, clonogenic, and multipotent and regenerate infarcted myocardium, improving cardiac function. Proceedings of the National Academy of Sciences of the United States of America 102: 8966–8971.
Matsuura, K., T. Nagai, N. Nishigaki, T. Oyama, J. Nishi, H. Wada, M. Sano, H. Toko, H. Akazawa, and T. Sato. 2004. Adult cardiac Sca-1-positive cells differentiate into beating cardiomyocytes. Journal of Biological Chemistry 279: 11384–11391.
Beltrami, A.P., Barlucchi, L., Torella, D., Baker, M., Limana, F., Chimenti, S., Kasahara, H., Rota, M., Musso, E., Urbanek, K., and other 4 authors. 2003. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114: 763–776.
Torella, D., C. Indolfi, D.F. Goldspink, and G.M. Ellison. 2008. Cardiac stem cell-based myocardial regeneration: towards a translational approach. Cardiovasc Hematol Agents Med Chem 6: 53–59.
Messina, E., L. De Angelis, G. Frati, S. Morrone, S. Chimenti, F. Fiordaliso, M. Salio, M. Battaglia, M.V.G. Latronico, and M. Coletta. 2004. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circulation research 95: 911–921.
Rossini, A., A. Zacheo, D. Mocini, P. Totta, A. Facchiano, R. Castoldi, P. Sordini, G. Pompilio, D. Abeni, M.C. Capogrossi, and A. Germani. 2008. HMGB1-stimulated human primary cardiac fibroblasts exert a paracrine action on human and murine cardiac stem cells. J Mol Cell Cardiol 44: 683–693.
Rossini, A., C. Frati, C. Lagrasta, G. Graiani, A. Scopece, S. Cavalli, E. Musso, M. Baccarin, M. Di Segni, and F. Fagnoni. 2011. Human cardiac and bone marrow stromal cells exhibit distinctive properties related to their origin. Cardiovascular research 89: 650–660.
Dawn, B., A.B. Stein, K. Urbanek, M. Rota, B. Whang, R. Rastaldo, D. Torella, X.L. Tang, A. Rezazadeh, and J. Kajstura. 2005. Cardiac stem cells delivered intravascularly traverse the vessel barrier, regenerate infarcted myocardium, and improve cardiac function. Proceedings of the National Academy of Sciences of the United States of America 102: 3766.
Torella, D., G.M. Ellison, S. Méndez-Ferrer, B. Ibanez, and B. Nadal-Ginard. 2006. Resident human cardiac stem cells: Role in cardiac cellular homeostasis and potential for myocardial regeneration. Nature Clinical Practice Cardiovascular Medicine 3: S8–S13.
Kuang, D., X. Zhao, G. Xiao, J. Ni, Y. Feng, R. Wu, and G. Wang. 2008. Stem cell factor/c-kit signaling mediated cardiac stem cell migration via activation of p38 MAPK. Basic research in cardiology 103: 265–273.
Hochman, J.S., and B.H. Bulkley. 1982. Expansion of acute myocardial infarction: an experimental study. Circulation 65: 1446–1450.
Bearzi, C., M. Rota, T. Hosoda, J. Tillmanns, A. Nascimbene, A. De Angelis, S. Yasuzawa-Amano, I. Trofimova, R.W. Siggins, and N. LeCapitaine. 2007. Human cardiac stem cells. Proceedings of the National Academy of Sciences 104: 14068.
Urbanek, K., D. Torella, F. Sheikh, A. De Angelis, D. Nurzynska, F. Silvestri, C.A. Beltrami, R. Bussani, A.P. Beltrami, and F. Quaini. 2005. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proceedings of the National Academy of Sciences of the United States of America 102: 8692.
Acknowledgments
This study was supported in part by a grant from the National Natural Science Foundation of China (to B.Y., Grant No. 30871064). The authors thank Baixiang Li, Wei Liu and Hulun Li for their helps with technical assistance, and also thank Dr. Meng Sun for assistance with the statistics.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Liu and Y. Wang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Liu, J., Wang, Y., Du, W. et al. Sca-1-Positive Cardiac Stem Cell migration in a Cardiac Infarction Model. Inflammation 36, 738–749 (2013). https://doi.org/10.1007/s10753-013-9600-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-013-9600-8