
Abstract
Objective
It was reported that there are cardiac stem cells (CSCs) in the rat heart, and they could reconstitute well-differentiated myocardium that are formed by blood-carrying new vessels and myocytes. However, how do the CSCs migrate into the peri-infarcted areas after myocardial infarction (MI)? It remains entirely unknown about the signal transduction involved in the migration of CSCs.
Methods and results
Rat heart MI was induced by left coronary artery ligation. Both immunohistochemical staining and Western blotting analysis was performed to detect the expression of SCF protein, and RT-PCR was conducted for the expression of SCF mRNA. Cardiac stem cells were isolated from rat hearts, and a cardiac stem cell migration assay was performed using a 48-well chemotaxis chamber system. On day 5 after MI in rats, the expression of stem cell factor (SCF) mRNA and protein was significantly increased in the peri-infarcted area, which was matched with more accumulation of CSCs in the region and improvement of cardiac function, which was blocked by p38 MAPK selective inhibitor SB203580. In in vitro experiments, SCF induced CSC migration in a concentration-dependent manner, and the antibody against SCF receptor (c-kit) blocked the SCF-induced CSC migration. Western blot analysis showed that the phosphorylated p38 MAPK (Phospho-p38 MAPK) was highly increased in the SCF-treated CSCs, and the inhibition of p38 MAPK activity significantly attenuated SCF-induced the migration of CSCs.
Conclusion
It demonstrated that SCF/c-kit signaling may mediate the migration of CSCs via activation of p38 MAPK.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Myocardial infarction (MI) is a leading cause of heart failure. Because of the ingrained concept of the myocardium as a terminally differentiated organ, it has been assumed that any attempt to replace the lost myocytes using cellular therapy would require the introduction of exogenous cells into the myocardium. However, it has recently been reported that the adult heart harbors primitive stem/progenitor cells [4, 7, 8, 18, 24]. Beltrami et al. [4] demonstrated that cardiac stem cells (CSCs) are a pure population of cloned c-kit-positive cells that can be differentiated into cardiomyocytes, smooth muscle cells and vascular endothelium. Previous studies have documented the ability of CSCs to regenerate infarcted myocardium when CSCs were injected intramyocardially in the per-infarct region after a permanent coronary occlusion. Dawn et al. [8] also reported that after intracoronary administration, CSCs could traverse the vessel barrier, regenerate infarcted myocardium, and improve cardiac function. Despite these encouraging observations, the mechanism of CSCs to repair the heart remains unclear. Especially, how do the CSCs migrate into the peri-infarcted areas when myocardial infarction occurs? Little has been reported on the migration of CSCs, and it remains entirely unknown about the signal transduction involved in the migration of CSCs.
Stem cell factor (SCF, also called c-kit ligand) is a critical factor for the development, survival, proliferation and migration of mast cells [12, 32]. By binding to c-kit, a tyrosine kinase-containing receptor, SCF can induce cell migration, adhesion to extracellualar matrix components and secretion of mediators [17, 39]. Our previous study has shown that SCF induced the bone marrow stem cell migration and improved the cardiac function [40]. However, whether SCF is involved in the cardiac stem cell migration remains unknown.
It was well known that the family of MAPKs plays a key role in cellular response to cytokine stimulation. The main extracellular signal-regulated kinase of MAPKs consists of ERK1/2, p38 MAPK and JNK. However, the JNK and p38 MAPK pathways have been reported to be responsible for the cytokine-induced signaling of cell migration [21].
In the present study, we reported the identification of SCF as a cytokine that is highly expressed in peri-infarcted areas after MI, and mediated the migration of CSCs via activation of p38 MAPK.
2 Materials and methods
2.1 Rat myocardial infarction model
A rat myocardial infarction model leading to left ventricle dysfunction was used in present study. Male Wistar rats (250–300 g) were randomized to coronary artery ligation or sham-operated groups. At day 5 after ligation, heart tissues were prepared for the detection of SCF mRNA and protein. All procedures were performed in accordance with the Guidelines of the Hubei Council of Animal Care and approved by the Animal Use Subcommittee at the Huazhong University of Science and Technology, China.
2.2 Hemodynamic measurement
Three weeks after coronary artery ligation or sham operation, rats were re-anesthetized with sodium pentobarbital. The right carotid artery was cannulated with a Millar microtip pressure transducer catheter. After arterial blood pressure and heart rate measurements were obtained, the catheter was advanced into the left ventricle (LV) for the measurement of LV systolic and end-diastolic pressures as well as the maximal rate of pressure development (+dP/dt max) and rate of relaxation (−dP/dt min) of LV.
2.3 Immunohistochemical staining
To detect the expression of SCF in the heart tissues, the rabbit antibody (1:100, Pepro Tech EC Ltd) against murine SCF was used. Endogenous peroxidase was blocked by 0.3% H2O2 for 20 min. The secondary antibody for immunostaining was biotin-conjugated anti-rabbit immunoglobulin (1:200, Dako).
2.4 Isolation and culture of CSCs from the adult rat heart
CSCs were isolated from the hearts of male Wistar rats (250–300 g) by a method described previously with a minor modification [4]. Briefly, the rat was injected with heparin (5,000 IU/kg, i.p.) 20 min prior to the experimental protocol, and then was killed by cervical dislocation. The heart was excised and the aorta was cannulated rapidly. The cannulated heart was mounted on a Langendorff perfusion apparatus with constant flow and perfusion pressure was monitored. The heart was firstly perfused with Ca2+-free Tyrode solution for 10 min to remove the blood and then digested by 0.5 mg/ml collagenase (Sigma) and 0.05 mg/ml trypsin (Difoo) at 37°C for 30 min. After the enzymatic digestion was terminated, the heart tissue was chopped and cell suspension collected was filtered with a strainer (Becton Dickson). Afterward, cells were incubated with a rabbit anti-c-kit antibody (Santa Cruz) and separated by using sheep anti-rabbit immunomagnetic microbeads (Dynal Biotech). Small round cells, containing most of the c-kitpos population, were separated. These c-kitpos cells were cultured for 3–5 days with Dulbecco’s MEM (Invitrogen) containing fetal calf serum, bFGF, and LIF at 37°C. After recovery, they were used for subsequent experiments.
2.5 Western immunoblotting analysis
The protein level of SCF in heart tissues was determined by Western immunoblotting analysis. Briefly, a total of 50 µg of protein in each sample were separated by SDS polyacrylamide gel (12.5%) electrophoresis followed by electrophoretic transfer of proteins from the gel to a nitrocellulose membrane. The membrane was probed with rabbit anti-SCF antibody (Santa Cruz Biotechnology). Bands corresponding to SCF protein were visualized using enhanced chemiluminescence reagents and analyzed with a gel documentation system. For the detection of phosphorylation of p38 MAPK, the anti-phosphotyrosin antibody (Cell Signaling Technology, Inc., USA) against P38 was used. To ensure similar amounts of proteins in each sample, the same membrane was stripped off, and reprobed with the antibodies against p38 MAPK.
2.6 Reverse transcription (RT)-PCR analysis
Total RNA was extracted from the left ventricle with Trizol reagent. RT-PCR was performed to analyze mRNA expression of SCF or c-kit with the primers and conditions as described previously [14, 41]. PCR products were separated on 1.5% of agarose gel.
2.7 Chemotaxis assay
Chemotaxis experiments were performed using a 48-well chemotaxis chamber technique (Neuro Probe) as previously described with a minor modification [29]. Briefly, 25 µl of medium (RPMI 1640, Gibco) alone or medium containing 5, 10, 30, 50, 100 ng/ml SCF (Santa Cruz) was placed in the lower chamber. A polycarbonate membrane with a 5-µm pore size separated the upper and lower chamber. CSCs resuspended in RPMI 1640 (50 µl) were placed in each well of the upper chamber. For inhibition experiment, CSCs were preincubated with these inhibitors for 20 min, and then added into the upper chamber. The chamber was then incubated for 3 h at 37°C in a humidified atmosphere with 5% CO2 and then disassembled. The membrane was removed and scraped to remove non-migrating CSCs from the upper surface. Then the membrane was fixed and stained. The numbers of CSCs that had migrated to the lower surface of the membrane were counted in ten random high-power fields (HPFs) by light microscopy, and a chemotactic index (CI) was calculated to express stimulated migration. Each assay was performed in triplicate wells.
2.8 Cell labeling and assessment of cell migration in vivo
To detect the response of CSCs to SCF in vivo, the cultured CSCs were labeled with the thymidine analogue 5-bromo-2′deoxyuridine (BrdU; Zymed) as described previously [3]. The labeled cells (1 × 105) were injected into AV-groove followed by a coronary ligation. For the inhibition experiment, the labeled cells were suspended in the medium containing 10 µM SB203580, then were injected into AV-groove. At day 5 after ligation, histological examination was performed in peri-infarcted regions to localize the BrdU-labeled cells which migrated from AV-groove after myocardial infarction by using mouse anti-BrdU (Zymed) antibody and fluorescent antibody (secondary antibody) in the heart. For the cardiac function, at three weeks, the hemodynamic parameters were measured.
2.9 Statistics analysis
All data are expressed as mean ± SEM. For analysis of differences between two groups, Student’s t test was performed. For multiple groups, ANOVA was carried out followed by Student–Newman–Keuls test. The level of statistical significance was set at P < 0.05.
3 Results
3.1 SCF expression in rat left ventricle myocardium after MI
The expression of SCF mRNA in LV myocardium was studied at day 5 after MI in rats. As shown in Fig. 1A, quantitative analysis of SCF mRNA expression to β-actin was illustrated. The result showed that the sham-operated heart tissue exhibited weak expression of SCF mRNA, however, after MI, SCF mRNA expression was significantly increased in the peri-infarcted area. To further confirm this result, we detected SCF expression at protein level by immunohistochemical staining and Western blotting analysis. The results showed that SCF protein expression was markedly increased in peri-infarcted area compared to the sham (Fig. 1B, C).

SCF expression in left ventricle tissues at day 5 following myocardial infarction in rats. A SCF mRNA expression by RT-PCR. B SCF protein expression by immunohistochemical staining (▼ indicates expression of SCF in peri-infarcted areas). C SCF protein levels by Western blot analysis in left ventricle tissues following myocardial infarction or sham operation. N = 5–7 per group. Results are mean ± SEM from four independent experiments. * Means P < 0.05 versus corresponding sham
3.2 CSC migration to the peri-infarcted regions after MI
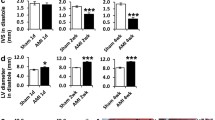
In order to understand whether the increased SCF expression in the peri-infarcted area led to more accumulation of CSCs after MI, the assessment of CSC migration in vivo was performed by using BrdU- labeled CSCs, which were injected into AV-groove. As shown as in Fig. 2A, the increased SCF expression was matched with more accumulation of CSCs in the peri-infarcted regions at 5 days after MI. The administration of SB203580 resulted in a significant reduction of the accumulation of CSCs in the peri-infarcted regions (Fig. 2B) at day 5 after MI, which demonstrated that p38 MAPK may play important roles in SCF-induced CSC migration.

The effect of SB203580 on the migration of BrdU labeled CSCs into the peri-infarcted regions. The migrated CSCs to the peri-infarcted areas was detected by using mouse anti-BrdU antibody and fluorescent-labeled antibody (secondary antibody) A, the BrdU-labeled cell was troponon-I positive (A d). The effect of SB203580 on the migration was assessed (B) at day 5 after MI, the administration of SB203580 was conducted to detect the blockage of SB203580 on the accumulation of CSCs in the peri-infarcted regions. The cardiac function was measured at 3 weeks after MI, the changes of LV +dP/dt max and −dP/dt min were shown in C and D, respectively. N = 7–9 per group * Means P < 0.05 versus corresponding sham; ** means P < 0.05 versus peri-infarct or MI; Δ means P < 0.05 versus group MI + C
3.3 The effect of SB203580 on cardiac function
In order to understand the functional relevance of the reduction of CSC migration into the peri-infarcted regions via the inhibition of p38 MAPK, the cardiac function was measured at 3 weeks after MI. Treatment with SB203580 significantly decreased LV +dP/dt and −dP/dt to a greater extent in the rats compared to no treatment after MI (Fig. 2C, D), however, there were no significant differences in heart rate, mean arterial pressure, or LV systolic pressure and LV end-diastolic pressure between SB203580-treated and non-treatment.
3.4 Features and identification of c-kitpos cells from the adult rat heart
By using immunomagnetic microbeads, the c-kitpos cells were isolated and collected from adult rat hearts [4]. This can finally result in the c-kitpos cells with a purity of 92%. Under light microscopy, they were small, round, phase-bright, and suspended in the medium. The expression of c-kit was detected by RT-PCR. As shown in Fig. 3A, c-kit mRNA was detectable in isolated CSCs, however, as a negative control, the NIH 3T3 cells cannot express c-kit mRNA. These results confirmed that these isolated cell were really c-kitpos CSCs.

CSC migration induced by SCF. CSCs were isolated from adult rat heart and RT-PCR analysis of c-kit mRNA was performed in the isolated cells A, NIH 3T3 cells were used as a negative control, and β-actin as an internal control for RT-PCR. In vitro CSC chemotaxsis assays were performed on a 48-well Boyden chamber system. The lower chamber was placed with medium (RPMI 1640) alone or medium containing 5, 10, 30, 50, 100 ng/ml SCF, while CSCs were added in the upper chamber. The medium alone was used in negative control experiments. Data are mean ± SEM, from four independent experiments. B Showed SCF-induced CSC migration with a dose dependent manner. C CSCs were pre-incubated with c-kit antibody (or IgG non-specific antibody) for 20 min, then were added into upper chamber for the migration assay (30 ng/ml of SCF). D The cells were pre-incubated with the P38 MAPK selective inhibitor SB203580 for 20 min, then added into upper chamber for the migration assay. * Means P < 0.05 versus corresponding control, ** means P < 0.05 versus corresponding SCF, ∇ means P > 0.05 versus corresponding SCF
3.5 Chemoattractant effects of SCF on CSC migration
During myocardial infarction, SCF was rapidly induced by the peri-infarcted area, and the increased SCF was matched with more accumulation of CSCs. We next asked if the SCF upregulation in the injured myocardium was functionally relevant for CSC migration. A Boyden chamber-based migration assay was established to quantitatively evaluate CSC migration in vitro. The lower chamber was placed with medium (RPMI 1640) alone or medium containing 5, 10, 30, 50, 100 ng/ml SCF, while the upper chamber was placed with CSCs. As shown in Fig. 3B, compared with the control group, the averaged numbers of migrated CSCs increased significantly as the concentration of SCF increased, which reach a peak at 30 ng/ml (n = 30, P < 0.05 compared with medium alone). As c-kit mRNA was not detectable in the NIH 3T3 cells [30], therefore we included NIH 3T3 cells in the Boyden chamber assay as a negative control, which did not induce migration of CSCs regardless of the concentration of SCF used. However, SCF resulted in a significant chemoattractant effect on CSCs in a dose dependent manner in vitro. This result documented that SCF could serve as a chemoattractant of CSCs in acute MI.
3.6 Role of c-kit antibody in SCF-induced CSC migration
SCF-induced CSC migration could be abolished by the pretreatment of the CSCs with c-kit blocking antibody, as shown in Fig. 3C. This migration inhibition did not occur after pretreatment with a control IgG
3.7 P38 MAPK was involved in the SCF-induced CSC migration
To explore whether SCF induced migration through the P38-dependent pathways in CSCs, the specific inhibitor SB203580, an agent that selectively inhibits p38 MAPK, was tested for the effect on SCF-induced CSC migration. CSCs were preincubated with SB203580 (10 µM) for 30 min, the cells were then added to the upper chambers for the CSC migration assay. As shown in Fig. 3D, application of the inhibitor significantly attenuated the effect of SCF on CSC migration. However, the number of migrated cells in the presence of inhibitor remains significantly greater than the control group which suggests that other signaling pathways may be involved in SCF-induced CSC migration. Western blot analysis showed that the levels of phospho-p38 MAPK were significantly increased in SCF-treated CSCs, however the total protein levels of p38 MAPK were not markedly altered in SCF-treated cells. The phospho-p38 MAPK reached a peak after a 15 min-incubation with SCF, and returned to basal level 60 min after application of SCF (Fig. 4).

Western blot analysis of P38 MAPK after SCF stimulation for 0–120 min in CSCs. A showed the original gels (P38 and phosphor- P38). B Showed the quantified data from three independent experiments. The results are mean ± SEM
4 Discussion
One of the contributing factors in the progression of heart failure is the loss of cardiomyocytes after myocardial infarction, combined with the absence of an adequate endogenous repair mechanism. A fibrous scar finally replaces the injured myocardium. The significance of cardiac c-kit positive cells has recently demonstrated that the result from Dr. Li’s lab showed that cardioprotective c-kit+ cells were from the bone marrow and regulated the myocardial balance of angiogenic cytokines in infarcted myocardium, thereby driving efficient cardiac repair [11]. Stem cell factor receptor induced progenitor and natural killer cell-mediated cardiac survival and repair after MI, which contributed to improved remodeling and cardiac function after MI [2]. Recently, stem cell based therapy has become a realistic option to replace damaged cardiomyocytes [13, 34]. Several cardiac stem/progenitor cells have been identified, which expressed the stem cell-related antigens c-kit, MDR1, Sca-1 or islet-1 [1, 4–7, 22]. In vitro data suggest that the growth potential of c-kit-positive CSCs is greater than that of others, although these cell categories give rise to all cardiac cell lineages [19]. The extraordinary clinical potential of myocardial regeneration makes the dissection of the biology of CSCs a challenging and exciting endeavor.
Since in fetal life, c-kit-positive cells colonize the yolk sack, liver, and probably other organs. The colonized organs express SCF, the ligand of the c-kit receptor [16, 33], so it was reasonable to assume that stem like cells are present in the heart from fetal life. Recently, CSC clusters have been found in the adult heart [5, 23, 37]. Although CSC clusters are scattered throughout the myocardium, they accumulate in the atria and apex, and are less numerous at the base and mid-portion of the left ventricle [1]. The cardiac niches are expected to control the physiological turnover of myocardial cells and the growth, migration, and commitment of primitive cells leaving the niches to replace old dying cells in the myocardium. However, it remains unknown how these CSCs migrate into the injured myocardium after MI. Anversa’s group has reported that HGF plays a critical role in migration and proliferation of c-kit positive CSC [36]. Our results showed an increased expression of SCF in the peri-infarcted area at the levels of mRNA and protein, which was matched with more accumulation of CSCs in the region. It drew the hypothesis that SCF may be involved in the activation of resident primitive c-kit-positive cells which migrate into the peri-infarcted area, thereby, in the increased formation of myocytes in the acutely infarcted heart, but it remains undetermined whether SCF could play a crucial role in this process.
For the purpose in the present study, the CSCs were isolated from heart tissues, and the migration assay was carried out by using 48-well chemotaxis chamber. Our data documented that SCF could attract migration of CSCs in a dose-dependent manner in vitro, which suggests that SCF may attract CSCs to injured myocardium and participate in the repair of heart in acute MI.
C-kit is a receptor tyrosine kinase (RTK), which constitutes a type III RTK subfamily with the receptors for platelet-derived growth factor (PDGF), colony-stimulating factor 1 (CSF-1), and flt-3 ligand [26, 43]. C-kit and its ligand stem cell factor (SCF) play an important role in hematopoiesis, malanogenesis, and gametogenesis [42]. Upon ligand stimulation, c-kit receptors dimerize, activate its intrinsic tyrosine kinase, and autophosphorylate. The phosphorylated c-kit receptor generates binding sites for SH2 domain-containing proteins, which include proteins of the p21 Ras-mitogen-activated protein kinase (MAPK) pathway [9], the p85 subunit of phosphatidylinositol 3′ kinase (PI3K) [28], phospholipase C-γ1, the Grb2 adaptor protein, the Src family kinases [20], Cbl, CRKL [27], p62Dok-1 [38], SHP1, and SHP2 [15]. Those proteins are subsequently activated or phosphorylated and further transduce signaling cascades that lead to various cellular responses. Migration is one of the unique and important cell functions undertaken by SCF/c-kit system. Much effort has been done to clarify signal transduction leading to SCF/c-kit-mediated proliferation and survival, but the signaling mechanism in SCF-mediated cell migration has not been clarified yet. Recently, a few reports indicated that JNK or p38 MAPK plays an important role in cell migration [21, 25, 31]. Shuji Ueda et al. [35] has provided evidence that SFK and PI3K cooperatively contribute to SCF-mediated cell migration through Ca2+ mobilization, and the signaling of SFK is transduced sequentially from p38 MAPK, Ca2+ influx, to Erk1/2. The JNK and p38 MAPK pathways have been reported to be responsible for the cytokine-induced signaling of cell migration [21]. We hypothesized that these signaling molecules may also play roles in SCF-mediated CSCs migration. To test the hypothesis, c-kit antibody and the specific inhibitors of JNK and p38 MAPK were used to block the SCF-induced migration of CSCs. Our present results showed that SCF-induced CSC migration was blocked by the antibody against c-kit, and significantly attenuated by a specific inhibitor of p38 MAPK in vivo and in vitro, and the functional relevance of the reduction of CSC migration into the peri-infarcted regions via the inhibition of p38 MAPK was assessed. The improvement of cardiac function after MI was significantly blocked by administration of p38 MAPK inhibitor. However, Engel et al. [10] reported that FGF1/p38 MAPK inhibitor therapy improves recovery after myocardial infarction, a long term observation on systemic administration of p38 MAPK inhibitor should be further carried out. Additionally, in the present study, the SCF-induced migration of CSCs cannot be influenced by specific inhibitor of JNK, although a higher concentration of JNK inhibitor was used (data not shown). However, the number of migrated cells in the presence of inhibitor remains significantly greater than the control group even if a higher concentration of SB203580 was used, which suggests that other signaling pathways may be involved in SCF-induced CSC migration. Further stimulation of CSCs with SCF markedly increased the phosphorylation of proteins of p38 MAPK, however the total protein levels of p38 MAPK did not change. It indicated that phosphorylation of proteins of p38 MAPK may be involved in SCF-induced migration of CSCs.
In summary, our study showed that myocardial infarction of rat heart led to an increased expression of SCF, which mediated migration of CSCs via stimulation of c-kit and activation of p38 MAPK.
References
Anversa P, Kajstura J, Nadal-Ginard B, Leri A (2003) Primitive cells and tissue regeneration. Circ Res 92:579–582
Ayach BB, Yoshimitsu M, Dawood F, Sun M, Arab S, Chen M, Higuchi K, Siatskas C, Lee P, Lim H, Zhang J, Cukerman E, Stanford W, Medin JA, Liu PP (2006) Stem cell factor receptor induces progenitor and natural killer cell-mediated cardiac survival and repair after myocardial infarction. Proc Natl Acad Sci USA 103:2304–2309
Barbash IM, Chouraqui P, Baron J, Feinberg MS, Etzion S, Tessone A, Miller L, Guetta E, Zipori D, Kedes LH, Kloner RA, Leor J (2003) Systemic delivery of bone marrow-derived mesenchymal stem cells to the infracted myocardium: feasibility, cell migration and body distribution. Circulation 108:863–868
Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A, Beltrami CA, Anversa P (2001) Evidence that human cardiac myocytes divide after myocardial infarction. N Engl J Med 344:1750–1757
Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Nadal-Ginard B, Anversa P (2003) Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114:763–776
Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S (2003) Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell 5:877–889
Chimenti C, Kajstura J, Torella D, Urbanek K, Heleniak H, Colussi C, Di Meglio F, Nadal-Ginard B, Frustaci A, Leri A, Maseri A, Anversa P (2003) Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res 93:604–613
Dawn B, Stein AB, Urbanek K, Rota M, Whang B, Rastaldo R, Torella D, Tang XL, Rezazadeh A, Kajstura J, Leri A, Hunt G, Varma J, Prabhu SD, Anversa P, Bolli R (2005) Cardiac stem cells delivered intravascularly traverse the vessel barrier, regenerate infarcted myocardium, and improve cardiac function. Proc Natl Acad Sci USA 102:3766–3771
Duronio V, Welham MJ, Abraham S, Dryden P, Schrader JW (1992) p21ras activation via hemopoietin receptors and c-kit requires tyrosine kinase activity but not tyrosine phosphorylation of p21ras GTPase-activating protein. Proc Natl Acad Sci USA 89:1587–1591
Engel FB, Hsieh PC, Lee RT, Keating MT (2006) FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proc Natl Acad Sci USA 103:15546–15551
Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P, Verma S, Weisel RD, Keating A, Li RK (2006) Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Invest 116:1865–1877
Galli SJ, Zsebo KM, Geissler EN (1994) The kit ligand, stem cell factor. Adv Immunol 55:1–96
Histov M, Weber C (2006) The therapeutic potential of progenitor cells in ischemic heart diseases—past, present and future. Basic Res Cardiol 101:1–7
Konard L, Munir Keilani M, Cordes A, Voulck-Badouin E, Laible L, Albrecht M, Renneberg N, Aumouller G (2005) Rat sertoli cells express epithelial but also mesenchymal genes after immortalization with SV40. Biochim Biophy Acta 1722:6–14
Kunisada T, Yoshida H, Yamakazi H, Miyamoto A, Hemmi H, Nishimura E, Shultz LD, Nishikawa S, Hayashi S (1998) Transgene expression of steel factor in the basal layer of epidermis promotes survival, proliferation, differentiation and migration of melanocyte precursors. Development 125: 2915–2293
Laflamme MA, Myerson D, Saffitz JE, Murry CE (2002) Evidence for cardiomyocyte repopulation by extracardiac progenitors in transplanted human hearts. Circ Res 90:634–640
Lam V, Kalesnikoff J, Lee CW, Hernandez-Hansen V, Wilson BS, Oliver JM, Krystal G (2003) IgE alone stimulates mast cell adhesion to fibronectin via pathways similar to those used by IgE + antigen but distinct from those used by Steel factor. Blood 102:1405–1413
Leri A, Kajstura J, Anversa P (2005) Cardiac stem cells and mechanisms of myocardial regeneration. Physiol Rev 5:1373–416
Linke A, Mueller P, Nurzynska D, Casarsa C, Torella D, Nascimbene A, Castaldo C, Cascapera S, Bohm M, Quaini F, Urbanek K, Leri A, Hintze TH, Kajstura J, Anversa P (2005) Cardiac stem cells in the dog heart regenerate infarcted myocardium improving cardiac performance. Proc Natl Acad Sci USA 102:8966–8971
Linnekin D, DeBerry CS, Mou S (1997) Lyn associates with the juxtamembrane region of c-Kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J Biol Chem 272:27450–27455
Lewis TS, Shapiro PS, Ahn NG (1998) Signal transduction through MAP kinase cascades. Adv Cancer Res 74:49–139
Lyngbaek S, Schneider M, Hansen JL, Sheikh SP (2007) Cardiac regeneration by resident stem and progenitor cells in the adult heart. Basic Res Cardiol 102:101–114
Nadal-Ginard B, Kajstura J, Anversa P, Leri A (2003) A matter of life and death: cardiac myocyte apoptosis and regeneration. J Clin Invest 111:1457–1459
Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, Entman ML, Schneider MD (2003) Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA 100:12313–12318
O’Laughlin-Bunner B, Radosevic N, Taylor ML, Shivakrupa, DeBerry C, Metcalfe DD, Zhou M, Lowell C, Linnekin D (2001) Lyn is required for normal stem cell factor-induced proliferation and chemotaxis of primary hematopoietic cells. Blood 98:343–350
Qiu FH, Ray P, Brown K, Barker PE, Jhanwar S, Ruddle FH, Besmer P (1988) Primary structure of c-kit: relationship with the CSF-1/PDGF receptor kinase family—oncogenic activation of v-kit involves deletion of extracellular domain and C terminus. EMBO J 7:1003–1011
Sattler M, Salgia R, Shrikhande G, Verma S, Pisick E, Prasad KV Griffin JD (1997) Steel factor induces tyrosine phosphorylation of CRKL and binding of CRKL to a complex containing ckit, phosphatidylinositol 3-kinase, and p120(CBL). J Biol Chem 272:10248–10253
Serve H, Hsu YC, Besmer P (1994) Tyrosine residue 719 of the c-kit receptor is essential for binding of the P85 subunit of phosphatidylinositol (PI) 3-kinase and for c-kit- associated PI 3-kinase activity in COS-1 cells. J Biol Chem 269:6026–6030
Song ZH, Zhong M (2000) CB1 cannabinoid receptor-mediated cell migration. J Pharmacol Exp Ther 294:204–209
Sun L, Lee J, Fine HA (2004) Neuronally expressed stem cell factor induces neural stem cell migration to areas of brain injury. J Clin Invest 113:1364–1374
Sundstrom M, Alfredsson J, Olsson N, Nilsson G (2001) Stem cell factor-induced migration of mast cells requires p38 mitogen-activated protein kinase activity. Exp Cell Res 267:144–151
Suto H, Nakae S, Kakurai M, Sedgwick JD, Tsai M, Galli SJ (2006) Mast cell-associated TNF promotes dendritic cell migration. J Immunol 176:4102–4112
Teyssier-Le Discorde M, Prost S, Nandrot E, Kirzenbaum M (1999) Spatial and temporal mapping of c-kit and its ligand, stem cell factor expression during human embryonic haemopoiesis. Br J Haematol 107:247–253
Templin C, Kotlarz D, Marquart F, Faulhaber J, Brendecke V, Schaefer A, Tsikas D, Bonda T, Hilfiker-Kleiner D, Ohl L, Naim HY, Foerster R, Drexler H, Limbourg FP (2006) Transcoronary delivery of bone marrow cells to the infarcted murine myocardium: feasibility, cellular kinetics, and improvement in cardiac function. Basic Res Cardiol 101:301–310
Ueda S, Mizuki M, Ikeda H, Tsujimura T, Matsumura I, Nakano K, Daino H, Honda Zi Z, Sonoyama J, Shibayama H, Sugahara H, Machii T, Kanakura Y (2002) Critical roles of c-Kit tyrosine residues 567 and 719 in stem cell factor-induced chemotaxis: contribution of src family kinase and PI3-kinase on calcium mobilization and cell migration. Blood 99:3342–3349
Urbanek K, Rota M, Cascapera S, Bearzi C, Nascimbene A, De Angelis A, Hosota T, Chimenti S, Baker M, Limana F, Nurzynska D, Torella D, Rotatori F, Rastaldo R, Musso E, Quaini F, Leri A, Kajstura J, Anversa P (2005) Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. Circ Res 97:663–673
Urbanek K, Torella D, Sheikh F, Nurzynska D, Silvestri F, Beltrami CA, Bussani R, Beltrami AP, Quaini F, Bolli R, Leri A, Kajstura J, Anversa P (2005) Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc Natl Acad Sci USA 102:8692–8697
van Dijk TB, van Den Akker E, Amelsvoort MP, Mano H, Lowenberg B, von Lindern M (2000) Stem cell factor induces phosphatidylinositol 3-kinase-dependent Lyn/Tec/Dok-1 complex formation in hematopoietic cells. Blood 96:3406–3413
Vosseller K, Stella G, Yee NS, Besmer P (1997) c-kit receptor signaling through its phosphatidylinositide-3′-kinase-binding site and protein kinase C: role in mast cell enhancement of degranulation, adhesion, and membrane ruffling. Mol Biol Cell 8:909–922
Wang G, Lei M, Lu X, Feng Q (2004) Bone marrow stem cell migration to the infracted myocardium: role of TNF. Faseb J 18:A1286
Wang R, Li J, Yashpal N (2004) Phenotypic analysis of c-kit expression in epithelial monolayers derived from postnatal rat pancreatic islets. J Endocrinol 182:113–122
Witte ON (1990) Steel locus defines new multipotent growth factor. Cell 63:5–6
Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U, Ullrich A (1987) Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J 6:3341–3351
Acknowledgements
This study was supported in part by research grants 30470710 from the National Natural Science Foundation of China, NCET-04-0711 from the Program for New Century Excellent Talents in University, and 2005ABB009 from the Natural Science Foundation of Hubei.
Author information
Authors and Affiliations
Corresponding author
Additional information
Returned for 1. Revision: 22 August 2007 1. Revision received: 11 October 2007
Returned for 2. Revision: 31 October 2007 2. Revision received: 2 November 2007
Rights and permissions
About this article
Cite this article
Kuang, D., Zhao, X., Xiao, G. et al. Stem cell factor/c-kit signaling mediated cardiac stem cell migration via activation of p38 MAPK. Basic Res Cardiol 103, 265–273 (2008). https://doi.org/10.1007/s00395-007-0690-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-007-0690-z