
Abstract
The study of glacial relict species has been focused on understanding how the biogeographic patterns of species have developed. A number of studies using phylogenetic and population genetics approaches have been conducted for terrestrial glacial relict species, and the mechanisms of their formation have been elucidated. On the other hand, less focus has been placed on glacial relict species inhabiting freshwater systems. In particular, stable lakes can serve as refugia during a glacial period, but research studies on freshwater relict species inhabiting lakes have not been well conducted. In order to clarify the mechanism of the glacial relict species in freshwater, we conducted a molecular phylogeny analysis, divergence time estimation, and a biogeographic reconstruction on freshwater Valvatidae molluscs, which have been considered as a glacial relict in the Japanese Archipelago. Our study shows that the valvatid fauna in the Japanese Archipelago was produced by multiple dispersal events from the Asian continent and by vicariance events during the period of the Pliocene–Quaternary glaciation. It includes multiple relict species that survived interglacial periods in different lakes. These findings suggest that the lakes can serve as refugia not only during glacial periods, but also during interglacial periods.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Global climate change, for example, cooling down in the last glacial period, has dramatic impacts on many biota and their distribution (e.g., Hewitt, 2004, 1996; Schmitt, 2007; Xu et al., 2009). One of the best biogeographic model cases for revealing the impact of such climate change are glacial relict species (Habel et al., 2010c). The glacial relict is a cold-adapted species or population, which had a widespread distribution in the last glacial period but now has a narrow geographic distribution in the current interglacial period (present) (Lomolino et al., 2006; Habel et al., 2010a). Historically, the formation mechanism of a glacial relict population by the glacial–interglacial cycle has been examined using the fossil record and distribution patterns (Coles & Colville, 1980; Hultén & Fries, 1986), but recent progress in molecular phylogeny and population genetics has addressed the specific time of establishment of disjunctive populations and distribution transitions (e.g., Habel et al., 2010b). As a result, many studies in Europe suggest that the formation of the glacial relict population is closely connected with the last glacial period (e.g., Schönswetter et al., 2003; Schmitt & Hewitt, 2006; Skrede et al., 2006; Schmitt et al., 2010). On the other hand, as research studies have progressed, it has also become clear that the relict species having similar distribution patterns seem to have different foundation mechanisms (e.g., Schmitt et al., 2006; Varga & Schmitt, 2008; Schmitt, 2009).
On the other hand, in the North and East Asian region where ice sheets were not formed even in the last glacial maximum (Ray & Adams, 2001), the more diverse formation mechanisms of glacial relicts are being unraveled as compared with Europe. For example, Baba et al. (2001, 2002) suggested that two relict bird species had immigrated in the last glacial period and became isolated in the Holocene. Also, in some species such as the Japanese shrew mouse, immigration occurred to the Japanese Archipelago in the last glacial period (Ohdachi et al., 2001). In contrast, cases of isolation and diversification on a long time scale were estimated in some Japanese alpine butterflies (Nakatani et al., 2007a, b, 2012). In addition, the various formation patterns, such as multiple immigration, have been clarified in alpine plants, which are the most well-studied examples of the glacial relict species in the Japanese Archipelago (Fujii et al., 1997, 1999; Senni et al., 2005; Fujii & Senni, 2006; Ikeda et al., 2006, 2008, 2009, 2012, 2014; Ikeda & Setoguchi, 2007, 2013). Although formation mechanisms of glacial relict species have been clarified, most of the research studies were focused on alpine relicts, and other relict species have not been treated satisfactorily. In particular, it has been pointed out that lakes and hot springs can be refugia both in the short and long term (Martens, 1997; Bolotov et al., 2017), but there are few research studies focused on freshwater relict species except for some crustaceans and fishes in high latitude regions, which can inhabit the sea (e.g., Väinölä et al., 2001; Kontula & Väinölä, 2003; Dooh et al., 2006; Sheldon et al., 2008). Therefore, we focused on Japanese valvatid snails to clarify the formation mechanisms of glacial relict species in freshwater systems.
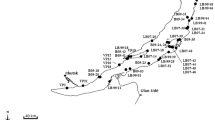
The family Valvatidae Gray, 1840 (Mollusca: Gastropoda), is a group of freshwater molluscs, which is distributed mainly in the high latitude area of the Northern Hemisphere (Strong et al., 2008; Haszprunar, 2014), and four species have been described in Japan (Habe, 1990; Masuda & Uchiyama, 2004). Of these four species, two are endemic to some restricted lakes (Fig. 1). Valvata biwaensis (Preston, 1916) is endemic to ancient Lake Biwa and V. kizakikoensis (Fujita & Habe, 1991) is also endemic to two connected lakes (Lake Kizaki and Lake Nakatsuna) (Fujita & Habe, 1991; Masuda & Uchiyama, 2004; Kihira et al., 2009). Of the remaining two species, V. japonica (Martens, 1877) has the widest distribution (Fig. 1), and it is distributed in the middle part of Honshu Island and further northwards including Hokkaido Island (Miyadi, 1935; Habe & Kawakami, 1982; Masuda & Uchiyama, 2004; Ito et al., 2005). V. hokkaidoensis Miyagi, 1935, is distributed only in Hokkaido and Aomori Prefecture, which is the northernmost prefecture in Honshu Island (Masuda & Uchiyama, 2004; Ito et al., 2005; Ohyagi, 2010a). Although the distribution of V. japonica is comparatively wide, the distribution in Honshu Island is discrete, and it is only recorded from Lake Nojiri, Lake Kawaguchi, Lake Ashinoko, the Sagami-gawa River drainage, and Lake Ogawara drainage (Habe & Kawakami, 1982; Kawabe et al., 2006; Ohyagi, 2010b). In addition, it is considered that of these locations in Honshu Island, the only collection records in recent years are from the Sagami-gawa River drainage and Lake Ogawara drainage, and valvatid molluscs of other localities may have become extinct or nearly extinct. Owing to this discrete distribution in Honshu Island, the valvatid molluscs in Honshu Island have been regarded as the relicts of the last glacial period (Habe & Kawakami, 1982). It is hypothesized that the Japanese valvatid species had expanded from the high latitude region to the south in the glacial period, and then they have been regressing during the interglacial period and now they are left behind in a stable lake with low water temperature. Although Habe & Kawakami (1982) concluded that valvatid species occupying Honshu Island were relicts of last glacial period because of their fragmented distribution patterns and age of the Japanese lakes except for Lake Biwa, the formation mechanisms of distinctive distribution (Fig. 1) and the origin of Japanese valvatid molluscs remain unclear.

A map of localities for the samples collected and distribution of Japanese Valvatidae. The bars indicate areas where the samples were collected. The circles and colors indicate the localities where Valvatidae is recorded. Gray color indicates situation nearly extinction or extinction (there is no record for more than about 20 years). The locality status in Hokkaido Island is not clear because of lack of investigation
Here, we conduct the molecular phylogeny of Japanese Valvatidae so as to address these unresolved issues. This attempt will be a good model case for not only clarifying the origin of relict valvatid mollusca, but also elucidating the formation mechanism of freshwater glacial relict species which has not been adequately studied. In addition, our research will help to clarify the formation of biota distributions in the Japanese Archipelago.
Materials and methods
Species sampling
We sampled nine described valvatid species, the sample comprising 20 individuals from 10 sites from the Japanese Archipelago, Russian Far East, Mongolia, and Lake Baikal for phylogenetic analysis. These samples included all four species described from Japan. Moreover, the sequence data of the world’s Valvatidae on GenBank are included for comparison as well with an ‘outgroup’ in phylogeny with reference to also Clewing et al. (2014). Collected Valvatidae species were identified by Masuda & Uchiyama (2004), Starobogatov et al. (2004), Kihira et al. (2009), Prozorova et al. (2009), and some original descriptions (Martens, 1877; Preston, 1916; Miyadi, 1935; Fujita & Habe, 1991). The genus name was provisionally treated as Valvata according to Clewing et al. (2014), because taxonomic problems of valvatid genus are not solved yet and are out of scope of the present research. Detailed information on the samples examined is given in Table 1 and in Fig. 1.
Molecular methods
DNA was isolated from individual gastropods, using the protocol of Sokolov (2000). The muscle tissue was homogenized in 800 μl of lysis buffer and incubated at 60°C for 1 h. Saturated potassium chloride (80 μl) was added to the lysate, and the solution was incubated for 5 min on ice before centrifugation for 10 min. The supernatant (700 μl) was poured into a new tube, extracted with a phenol/chloroform solution, and precipitated with an equal volume of 2-propanol. The DNA pellet was rinsed with 70% ethanol, dried, and dissolved in 50 μl of TE buffer.
To estimate the phylogenetic positions of North and East Asian Valvatidae, we sequenced fragments of the mitochondrial cytochrome oxidase subunit 1 (CO1), mitochondrial large ribosomal subunit (16S) as well as nuclear large ribosomal subunit (28S). The polymerase chain reaction (PCR) and primers are listed in Table 2. The PCR products were purified with Exo-SAP-IT (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK). Sequencing was performed using a BigDye™ terminator cycle sequencing ready reaction kit (Applied Biosystems, Foster City, CA, USA) and electrophoresed using an ABI 3130xl sequencer (Applied Biosystems, Carlsbad, CA, USA). The obtained CO1, 16S, and 28S sequences have been deposited in the DDBJ/EMBL/GenBank database (Table 1).
Phylogenetic analyses
There was no gap in the aligned sequences of CO1, and the 16S and 28S sequences were aligned with MUSCLE v3.8 (Edgar, 2004). To eliminate uncertainty of the alignment in this region, trimAl 1.2 (Capella-Gutiérrez et al., 2009) was used to select regions of the aligned sequences for analysis (Online Resources 1, 16S and 2, 28S). First, phylogenetic relationship was estimated for each locus. The phylogenetic trees were obtained using the Bayesian inference (BI) and maximum likelihood (ML) methods. Prior to the BI and ML analyses, we used the program Kakusan4-4.0.2011.05.28 (Tanabe, 2011) to select the appropriate models of sequence evolution (Table 3). Based on these models, ML analysis was performed with 500 iterations of the likely ratchet (Nixon, 1999; Vos, 2003) using Treefinder (Jobb, 2008) and Phylogears2, v2.2.2012.02.13 (Tanabe, 2012) software. For the ML analyses, we assessed nodal support by performing bootstrap analyses with 1,000 replications. The BI analysis was performed using MrBayes v3.1.2 (Ronquist & Huelsenbeck, 2003), with two simultaneous runs. Each run consisted of four simultaneous chains for two million generations and sampling of trees every 100 generations. We discarded the first 2,001 trees as burn-in after examining convergence and effective sample size (ESS) by Tracer v. 1.6 (Rambaut et al., 2013), so the remaining samples were used to estimate phylogeny. Then, topologies of these single locus trees (Online Resources 3–5) were exanimated. As a result, there was no major inconsistency among the analyzed sequences in supported tree topology of three loci, with the proviso that the tree of 28S had low resolution. Next, phylogenies were estimated by using combined locus. The same protocols as single locus analysis were used in combined locus analysis. The selected model was also shown in Table 3. The ML analysis was performed with 500 iterations of the likely ratchet and 1,000 replications of bootstrap analyses. The two simultaneous runs in BI analysis consisted of four simultaneous chains for 3,000,000 generations and sampling of trees every 100 generations. We discarded the first 3,001 trees as burn-in after checking by Tracer v. 1.6, so the remaining samples were used to estimate phylogeny.
Divergence time estimation
Estimation of divergence times was performed in BEAST2 v. 2.4.4 (Bouckaert et al., 2014) using the same dataset with the following settings: tree prior = Yule process, ngen = 20,000,000, samplefreq = 1,000. The two different clock models (strict clock and uncorrelated lognormal relaxed clock) were adapted for comparison. Substitution models of each partition were set as follows: CO1 = HKY + I + Γ, 16S = HKY + Γ, 28S = GTR + Γ. These models were selected using Kakusan4-4.0.2011.05.28 (Tanabe, 2011) and those available in BEAST2 v. 2.4.4 (Bouckaert et al., 2014). The model of CO1 was chosen from models in which the molecular clock rate is being considered in Wilke et al. (2009). A molecular clock rate (uniform prior) ranging from 0.0126 to 0.0189 (substitutions per site and Ma) was proposed for the COI gene for different Protostomia groups using the substitution model: HKY + I + Γ (see Wilke et al., 2009). Maximum clade credibility files were annotated in TreeAnnotator v. 2.4.4 (BEAST package; burn-in = 10%) summarizing the entire posterior distribution including a total of 20,001 trees after log and tree files were checked by Tracer v. 1.6.
Biogeographic analyses
To reconstruct ancestral areas and the likelihood of dispersal and vicariance events, we used Bayes–Lagrange (statistical dispersal–extinction–cladogenesis, S-DEC; Beaulieu et al., 2013) and statistical dispersal–vicariance analysis (S-DIVA; Yu et al., 2010) in RASP 3.2 (Yu et al., 2015). For these analyses, we used the same 20,001 trees generated from divergence time analysis using strict clock model with BEAST2 v. 2.4.4 (see above). The first 2001 trees of those were discarded on RASP 3.2. In addition, as a condensed tree, we used also the same maximum clade credibility tree using strict clock model generated from TreeAnnotator v. 2.4.4. Under both models, the outgroup was removed before the analyses. The following nine areas were defined: (A) Nearctic, (B) Europe and Turkey, (C) North Asia, (D) Lake Baikal, (E) East Asia excluding Japanese Archipelago, (F) Tibetan Plateau, (G) Honshu Island, (H) Hokkaido Island, (I) Lake Biwa. The boundary of areas was decided for convenience considering the biogeographic area and the ancient lakes (for detailed region, see Fig. 3). The S-DIVA models were calculated with default settings and max areas = 2, allowing for extinctions. The S-DEC analyses were run with default settings and max areas = 2.
Results
Phylogenetic relationships
For the molecular phylogenetic analyses, the ESS values visualized in Tracer v. 1.6 were considerably greater than 200. The inferred Bayesian phylogenetic relationships are shown in Fig. 2. Both ML and BI trees resulted in nearly identical topologies. The valvatid species formed six major clades and these were named as “I-VI.” The monophyly of five clades, excluding clade III, were strongly supported by both trees [Bayesian posterior probabilities (BPP) = 1.00, ML bootstrap value (BV) ≥ 76%]. In regard to clade III, the monophyly was also well supported in ML method (BV = 78%) in spite of BPP = 0.90. In addition, clades I–IV and V–VI formed deeper monophyletic clades, respectively. These two upper clades were also well supported by both methods.

The Bayesian phylogenetic tree inferred from a combined dataset of mtDNA and nDNA sequences (CO1, 16S, and 28S; 1,795 bp). Cornirostra pellucida (Laseron, 1954) is the outgroup chosen for the tree root. Each number at the terminal branch of the tree indicates the sample number and species name (Table 1). Numbers at the branch nodes represent BPP and BV. On the right side, vertical bars indicate nominal monophyletic clades
All Japanese materials were included in clades III and IV. These clades also contained materials from Russian Far East, Mongolia, China, and Siberia. Clade I was composed of Nearctic materials. Clade II species were mainly found in the materials from Europe, but also those from Tibetan Plateau, Turkey, and Western Siberia. Clade V was found in the materials from Europe, and clade IV from Lake Baikal. Materials of the same localities of the Japanese valvatid species showed monophyletic relationships except for two individuals from the Sagami River (Va85 and Va86). However, these two individuals were closest to V. kizakikoensis from Lake Nakatsuna of the Honshu Island, geographically closest area to the Sagami River. In clade IV, V. hokkaidoensis from Honshu Island and Hokkaido Island did not show a monophyletic relationship. The materials from these localities were phylogenetically closest to the continental valvatid materials. In clade III, materials of V. japonica were not monophyletic, and V. biwaensis from the Lake Biwa was the most ancestral lineage within the clade.
Divergence time estimation and biogeographic analyses
For the molecular clock analyses, the ESS values visualized in Tracer v. 1.6 were considerably higher than 200 in each analysis. The inferred Bayesian phylogenetic relationships using BEAST2 adopted strict clock model are shown in Fig. 3. We numbered the cardinal nodes from 1 to 17 for convenience. The topology of the tree was consistent with those obtained in MrBayes and Treefinder analyses (Fig. 2). Especially, the topology of the tree except for the low-supported clades was perfectly consistent. This result was also consistent with phylogenetic tree obtained by uncorrelated lognormal relaxed clock model (not shown). The mean time values of first divergence of two valvatid clades including Japanese materials (nodes 5 and 10) were estimated to be 1.97 and 1.17 Ma. Furthermore, in these major two clades, the mean divergence time of well-supported nodes excluding same locality clades (nodes 12–15, and 17) were, respectively, 0.86, 0.73, 0.58, 0.45, 0.20 Ma. The 95% HPD intervals of these nodes were in the Pleistocene (≥ 2.58 Ma). In addition, using uncorrelated lognormal relaxed clock model analyses, the mean divergence among these nodes occurred during the Pleistocene. The lower 95% HPD interval of newest node between two localities in Honshu Islands (node 17) were 0.10 Ma. In the uncorrelated lognormal relaxed clock model, this value was nearly same (0.11 Ma). The other detailed information is given in Table 4.

Maximum clade credibility tree generated by the BEAST2 analysis from the mtDNA and nDNA sequences (CO1, 16S, and 28S; 1,795 bp). The outgroup Cornirostra pellucida was not shown. On the left side, BPP and the divergence time are shown. Node bars indicate 95% CI of the divergence time by SC model analysis. The principal nodes are named nominal number. Red line indicates 2.75 Ma in which Northern Hemisphere glaciation started (Ravelo et al., 2004). In the right side, the results of two biogeographic analyses by RASP are shown. The circles beside the branch indicate the estimated ancestral region and biogeographic events. The striped color in the circles indicate connected region. Upper is the result by S-DEC method, lower is the result by S-DIVA method. The color corresponds to the distinguished region by author. The detailed information is given in the box in the upper right
Two different analyses using RASP estimated historic biogeographic area and events (Fig. 3). The valvatids occupied the region where North Asia (Region C) and two regions in Japanese Archipelago (Regions H and I) were combined. As for nodes 10, 13, 15, and 16, the results of the two analyses nearly agree with each other, and the ancestral regions of CI, CG, CH, CG were estimated, respectively. In addition, in both analyses, it was estimated that the ancestral region of node 5 was also CE in spite of three-quarters proportion in S-DEC analysis. Also, for all of these nodes, the event of vicariance was estimated by both analyses. For node 14, ancestral region CG was estimated by S-DEC, but S-DIVA estimated CG and G about half a possibility and dispersal event were estimated by both analyses. The ancestral region of node 12 was estimated to be C with a possibility of about three-quarters by S-DEC and half by S-DIVA and dispersal event were also estimated by both analyses. Lastly, the estimated principal ancestral region of the nodes 4 and 8 by both analyses was C. In these two nodes, the dispersal event was estimated. The detailed information is given in Table 4.
Discussion
There were two main clades of valvatids uncovered in our analyses. One clade represented Europe (clade V) and Lake Baikal (clade VI) and the other one represented the Nearctic (clade I), Eurasia (clade II), and the Russian Far East, Siberia, China, Mongolia, and the Japanese Archipelago (clades III and IV). Clade IV contained materials from Russian Far East, China, and the Japanese Archipelago but this lineage may be distributed widely in Eurasia because of the distribution area of Valvata sibirica (Middendorff, 1851) or V. frigida (Westerlund, 1873) (Vinarski & Kantor, 2016). Clades III and IV seem to indicate two separate origins for the Japanese valvatid populations as uncovered by our combined analyses. Also inside clades III and IV, the inferred phylogenetic relationships and results of RASP analyses suggest that valvatid fauna on the Japanese Archipelago have been produced by multiple events of dispersal from the continent followed by geographic isolation. The 95% CI of the estimated divergence time of BEAST2 is 1.26–2.04 Ma (even uncorrelated lognormal relaxed clock model; 1.11–3.27 Ma) even in the oldest node (node 5, but BPPs is not sufficient). This means that separation of the Japanese and continent lineages seemingly occurred after the late Pliocene or early Pleistocene (Fig. 3). Although further population genetic research studies are necessary to estimate the time when these respective vicariances and dispersals occurred, dispersal from continent to Japan followed by vicariance has occurred repeatedly.
It is estimated that the divergence of the six valvatid clades occurred after 2.75 Ma in both clock models (Fig. 3; Table 4). The Northern Hemisphere glaciations of the Pliocene–Quaternary started from 2.75 Ma (Ravelo et al., 2004), and the average temperature declined globally (Quante, 2010). This cooling has also occurred around the Japanese Archipelago (Hyodo & Kitaba, 2012). Because of the onset of glacial periods after the Pliocene, expansion of distribution range and diversification of those Valvatidae that adapted to cool environments have seemingly started, resulting in migration to the Japanese Archipelago from the continent. Referring to the geologic history of the Japanese Archipelago, it has repeatedly been connected to the continent during the periods of glaciation of Pleistocene (Ono, 1990; Kitamura & Kimoto, 2004, 2006), and glacial periods and interglacial periods have occurred repeatedly with a span of 41,000–100,000 years (Lisiecki & Raymo, 2007). This geologic history is consistent with our hypothesis.
The present discrete distribution and regional monophyletic pattern of Japanese valvatid molluscus may reflect their evolutionary history. Populations of Valvatidae within the Japanese Archipelago may have repeatedly expanded during the glacial periods and have been geographically isolated during interglacial periods when population ranges contracted. During the periods of range contraction, the lakes in Japan potentially provided refugia, because water temperature of the lakes is relatively stable and low due to existence of spring water and large water depth in general. Consequently, the populations are likely to have persisted mainly in those lakes where the water temperature was relatively stable and low during interglacial periods as well as at the present time. In fact, valvatid molluscs on the Honshu Island have been recorded only from the lakes or the rivers connected to lakes (Fig. 1) despite their wide distribution in high latitude areas of Japan. The latest separation event among the valvatid species in Honshu Islands is estimated at 0.10–0.30 Ma (uncorrelated lognormal relaxed clock model: 0.11–0.68 Ma). This upper 95% CI is even older than the last glacial period, suggesting that geographic isolations appear to have occurred before the formation of the lakes, and the lakes served as refugia during the last interglacial period. Some lakes that have valvatid molluscs were formed in relatively recent periods (Nitobe, 1975; Kobayashi, 2008), supporting this hypothesis.
Traditionally, valvatid molluscs in Japan have been considered to be relicts of last glacial period (Habe & Kawakami, 1982). However, our results suggest that they have complicated histories, such as multiple immigrations from the continent and separation of distributions by evacuation to the lakes, though further research studies of population genetics are required. The present findings suggest that the lakes may serve as not only refugia for temperate-adapted taxa during glacial periods (Bolotov et al., 2017; Liang et al., 2017) but also refugia for cold-adapted taxa during interglacial periods.
References
Baba, Y., Y. Fujimaki, R. Yoshii & H. Koike, 2001. Genetic variability in the mitochondrial control region of the Japanese rock ptarmigan Lagopus mutus japonicus. Japanese Journal of Ornithology 50: 53–64.
Baba, Y., Y. Fujimaki, S. Klaus, O. Butorina, S. Drovetskii & H. Koike, 2002. Molecular population phylogeny of the hazel grouse Bonasa bonasia in East Asia inferred from mitochondrial control-region sequences. Wildlife Biology 8: 251–259.
Beaulieu, J. M., D. C. Tank & M. J. Donoghue, 2013. A Southern Hemisphere origin for campanulid angiosperms, with traces of the break-up of Gondwana. BMC Evolutionary Biology 13: 80.
Bolotov, I. N., O. V. Aksenova, Y. V. Bespalaya, M. Y. Gofarov, A. V. Kondakov, I. S. Paltser, A. Stefansson, O. V. Travina & M. V. Vinarski, 2017. Origin of a divergent mtDNA lineage of a freshwater snail species, Radix balthica, in Iceland: cryptic glacial refugia or a postglacial founder event? Hydrobiologia 787: 73–98.
Bouckaert, R., J. Heled, D. Kühnert, T. G. Vaughan, C. H. Wu, D. Xie, M. A. Suchard, A. Rambaut & A. J. Drummond, 2014. BEAST2: a software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10: e1003537.
Capella-Gutiérrez, S., J. M. Silla-Martínez & T. Gabaldón, 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25: 1972–1973.
Clewing, C., P. V. von Oheimb, M. Vinarski, T. Wilke & C. Albrecht, 2014. Freshwater mollusc diversity at the roof of the world: phylogenetic and biogeographical affinities of Tibetan Plateau Valvata. Journal of Molluscan Studies 80: 452–455.
Coles, B. & B. Colville, 1980. A glacial relict mollusc. Nature 286: 761.
Dooh, R. T., S. J. Adamowicz & P. D. H. Hebert, 2006. Comparative phylogeography of two North American ‘glacial relict’ crustaceans. Molecular Ecology 15: 4459–4475.
Edgar, R. C., 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32: 1792–1797.
Folmer, O., M. Black, W. Hoeh, R. A. Lutz & R. Vrijenhoek, 1994. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology 3: 294–2998.
Fujii, N. & K. Senni, 2006. Phylogeography of Japanese alpine plants: biogeographic importance of alpine region of central Honshu in Japan. Taxon 55: 43–52.
Fujii, N., K. Ueda, Y. Watano & T. Shimizu, 1997. Intraspecific sequence variation of chloroplast DNA in Pedicularis chamissonis Steven (Scrophulariaceae) and geographic structuring of the Japanese ‘Alpine’ plants. Journal of Plant Research 110: 195–207.
Fujii, N., K. Ueda, Y. Watano & T. Shimizu, 1999. Further analysis of intraspecific sequence variation of chloroplast DNA in Primula cuneifolia Ledeb. (Primulaceae): implication for biogeography of the Japanese alpine flora. Journal of Plant Research 112: 87–95.
Fujita, T. & T. Habe, 1991. Cincinna kizakikoensis n. sp. of the Family Valvatidae from Lakes Kizaki and Nakatsuna, Nagano Prefecture, Japan. Venus 50: 23–26.
Glöer, P., 2002. Süßwassergastropoden Nord- und Mitteleuropas. Bestimmungsschluüssel, Lebensweise, Verbreitung. ConchBooks, Hackenheim.
Habe, T., 1990. The list of Japanese freshwater mollusks:1. Hitachiobi 54: 3–6.
Habe, T. & A. Kawakami, 1982. Glacial relic species, Cincinna piscinalis japonica (Martens). Chiribotan 13: 64–65.
Habel, J. C., T. Assmann, T. Schmitt & J. C. Avise, 2010a. Relict species: from past to future. In Habel, J. C. & T. Assmann (eds), Relict Species Phylogeography and Conservation Biology. Springer, Berlin: 1–5.
Habel, J. C., C. Drees, T. Schmitt & T. Assmann, 2010b. Review refugial areas and postglacial colonizations in the Western Palearctic. In Habel, J. C. & T. Assmann (eds), Relict Species Phylogeography and Conservation Biology. Springer, Berlin: 189–197.
Habel, J. C., T. Schmitt & T. Assmann, 2010c. Relict species research: some concluding remarks. In Habel, J. C. & T. Assmann (eds), Relict Species Phylogeography and Conservation Biology. Springer, Berlin: 441–442.
Haszprunar, G., 2014. A nomenclator of extant and fossil taxa of the Valvatidae (Gastropoda, Ectobranchia). ZooKeys 377: 1–172.
Hauswald, A. K., C. Albrecht & T. Wilke, 2008. Testing two contrasting evolutionary patterns in ancient lakes: species flock versus species scatter in valvatid gastropods of Lake Ohrid. Hydrobiologia 615: 169–179.
Hewitt, G. M., 1996. Some genetic consequences of ice-ages, and their role in divergence and speciation. Biological Journal of the Linnean Society 58: 247–276.
Hewitt, G. M., 2004. Genetic consequences of climatic oscillations in the Quaternary. Philosophical Transactions of the Royal Society B 359: 183–195.
Hultén, E. & M. Fries, 1986. Atlas of North European Vascular Plants North of the Tropic of Cancer 1–3. Koeltz Scientific Books, Königstein.
Hyodo, M. & I. Kitaba, 2012. Late Pliocene magneto- and climatostratigraphic evolution toward the onset of the Quaternary in East Asia. The Journal of the Geological Society of Japan 118: 74–86.
Ikeda, H. & H. Setoguchi, 2007. Phylogeography and refugia of the Japanese endemic alpine plant, Phyllodoce nipponica Makino (Ericaceae). Journal of Biogeography 34: 169–176.
Ikeda, H. & H. Setoguchi, 2013. A multilocus sequencing approach reveals the cryptic phylogeographical history of Phyllodoce nipponica Makino (Ericaceae). Biological Journal of the Linnean Society 110: 214–226.
Ikeda, H., K. Senni, N. Fujii & H. Setoguchi, 2006. Refugia of Potentilla matsumurae (Rosaceae) located at high mountains in the Japanese Archipelago. Molecular Ecology 15: 3731–3740.
Ikeda, H., K. Senni, N. Fujii & H. Setoguchi, 2008. Survival and genetic divergence of an arctic-alpine plant, Diapensia lapponica subsp. obovata (Fr. Schm.) Hultén (Diapensiaceae), in the high mountains of central Japan during climatic oscillations. Plant Systematics and Evolution 272: 197–210.
Ikeda, H., K. Senni, N. Fujii & H. Setoguchi, 2009. High mountains of the Japanese Archipelago as refugia for arctic-alpine plants: phylogeography of Loiseleuria procumbens (L.) Desvaux (Ericaceae). Biological Journal of the Linnean Society 97: 403–412.
Ikeda, H., T. Carlsen, N. Fujii, C. Brochmann & H. Setoguchi, 2012. Pleistocene climatic oscillations and the speciation history of an alpine endemic and a widespread arctic-alpine plant. New Phytologist 194: 583–594.
Ikeda, H., V. Yakubov, V. Barkalov & H. Setoguchi, 2014. Molecular evidence for ancient relicts of arctic-alpine plants in East Asia. New Phytologist 203: 980–988.
Ito, T., A. Ohtaka, R. Ueno, Y. Kuwahara, H. Ubukata, S. Hori, T. Itoh, S. Hiruta, K. Tomikawa, N. Matsumoto, S. Kitaoka, S. Togashi, I. Wakana & A. Ohkawa, 2005. Aquatic macroinvertebrate fauna in Lake Takkobu, Kushiro Marsh, northern Japan. Japanese Journal of Limnology 66: 117–128.
Jobb, G., 2008. TREEFINDER version of October 2008 [available on internet at http://www.treefinder.de].
Kawabe, K., T. Sonohara, T. Yoshida & I. Aihara, 2006. Cincinna japonica (Gastropoda: Valvatidae) collected from a water reservoir on the Sagami River, Kanagawa Prefecture, Japan. Chiribotan 36: 116–118.
Kihira, H., O. Masuda & R. Uchiyama, 2009. Freshwater mollusks of Japan 1: freshwater mollusks of Lake Biwa and Yodogawa River. In Pisces, Revised Edition, Kanagawa.
Kitamura, A. & K. Kimoto, 2004. Reconstruction of the Southern Channel of the Japan Sea at 3.9–1.0 Ma. The Quaternary Research 43: 417–434.
Kitamura, A. & K. Kimoto, 2006. History of the inflow of the warm Tsushima Current into the Sea of Japan between 3.5 and 0.8 Ma. Palaeogeography, Palaeoclimatology, Palaeoecology 236: 355–366.
Kobayashi, M., 2008. Eruption history of the Hakone Central Cone Volcanoes, and geographical development closely related to eruptive activity in the Hakone Caldera. Research Report of Kanagawa Prefectural Museum Natural History 13: 43–60.
Kontula, T. & R. Väinölä, 2003. Relationships of Palearctic and Nearctic ‘glacial relict’ Myoxocephalus sculpins from mitochondrial DNA data. Molecular Ecology 12: 3179–3184.
Liang, Y., D. He, Y. Jia, H. Sun & Y. Chen, 2017. Phylogeographic studies of schizothoracine fishes on the central Qinghai–Tibet Plateau reveal the highest known glacial microrefugia. Scientific Reports 7: 10983.
Lisiecki, L. E. & M. E. Raymo, 2007. Plio–Pleistocene climate evolution: trends and transitions in glacial cycle dynamics. Quaternary Science Reviews 26: 56–69.
Lomolino, M. V., B. R. Riddle & J. H. Brown, 2006. Biogeography. Sinauer, Sunderland.
Martens, K., 1997. Speciation in ancient lakes. Trends in Ecology and Evolution 12: 177–182.
Masuda, O. & R. Uchiyama, 2004. Freshwater mollusks of Japan 2: freshwater mollusks of Japan. Including brackish water species. In Pisces, Tokyo.
Miller, M. P., D. E. Weigel & K. E. Mock, 2006. Patterns of genetic structure in the endangered aquatic gastropod Valvata utahensis (Mollusca: Valvatidae) at small and large spatial scales. Freshwater Biology 51: 2362–2375.
Miyadi, D., 1935. Descriptions of three new subspecies of Valvata from Nippon. Venus 5: 59–62, Pl. 3.
Nakatani, T., S. Usami & T. Itoh, 2007a. Phylogeographic history of the Japanese Alpine Ringlet Erebia niphonica (Lepidoptera, Nymphalidae): fragmentation and secondary contact. Transactions of the Lepidopterological Society of Japan 58: 253–275.
Nakatani, T., S. Usami & T. Itoh, 2007b. Molecular phylogenetic analysis of the Erebia aethiops groups (Lepidoptera, Nymphalidae). Transactions of the Lepidopterological Society of Japan 58: 387–404.
Nakatani, T., S. Usami & T. Itoh, 2012. Historic cycles of fragmentation and expansion in the Alpine butterfly Erebia ligea (Lepidoptera, Nymphalidae) on the Japanese Archipelago, inferred from mitochondrial DNA. Lepidoptera Science 63: 204–216.
Nitobe, K., 1975. The geomorphic history of Lake Ogawara. Annals of the Tohoku Geographical Assosiation 27: 25–35.
Nixon, K. C., 1999. The parsimony ratchet: a new method for rapid parsimony analysis. Cladistics 15: 407–414.
Ohdachi, S., N. E. Dokuchaev, M. Hasegawa & R. Masuda, 2001. Intraspecific phylogeny and geographical variation of six species of northeastern Asiatic Sorex shrews based on the mitochondrial cytochrome b sequences. Molecular Ecology 10: 2199–2213.
Ohyagi, A., 2010a. Valvata hokkaidoensis. In Aomori Prefecture Red Data Book Revision and Review Board and Nature Conservation Division in Aomori Prefecture (eds), The Rare Wildlife in Aomori Prefecture – Aomori Prefecture Red Data Book (2010 Revised Edition). Aomori Prefecture, Aomori: 311.
Ohyagi, A. 2010b. Cincinna japonica. In Aomori Prefecture Red Data Book Revision and Review Board and Nature Conservation Division in Aomori Prefecture (eds), The Rare Wildlife in Aomori Prefecture – Aomori Prefecture Red Data Book (2010 Revised Edition). Aomori Prefecture, Aomori: 311.
Ono, Y., 1990. The Northern Landbridge of Japan. The Quaternary Research 20: 183–192.
Palumbi, S. R., A. Martin, S. Romano, W. O. Mcmillan, L. Stice & G. Grabowski, 1991. The Simple Fool’s Guide to PCR. University of Hawaii Press, Honolulu.
Park, J. L., Ó. Foighil & D., 2000. Sphaeriid and Corbiculid clams represent separate heterodont bivalve radiations into freshwater environments. Molecular Phylogenetics and Evolution 14: 75–88.
Preston, H. B. 1916. Description of new freshwater shells from Japan. The Annals and Magazine of Natural History; Zoology, Botany, and Geology Being a Continuation of the Annals Combined with Loudon and Charlesworth’s Magazine of Natural History 8th Series 17: 159–163, Pl. 9.
Prozorova, L. A., T. Ya. Sitnikova, M. O. Zasypkina, P. O. Matafonov & A. Dulmaa, 2009. Freshwater Gastropoda in the Basin of Lake Baikal and adjacent territories. In Timoshkin, O. A. (ed.), Index of Animal Species Inhabiting Lake Baikal and Its Catchment Area. Basins and Channels in the South of East Siberia and North Mongolia, Book 1, Part 1, Vol. 2. Nauka, Novosibirsk: 170–188.
Quante, M., 2010. The changing climate: past, present, future. In Habel, J. C. & T. Assmann (eds), Relict Species Phylogeography and Conservation Biology. Springer, Berlin: 9–56.
Rambaut, A., A. J. Drummond & M. Suchard, 2013. Tracer v1.6 [available on internet at http://tree.bio.ed.ac.uk/software/tracer/].
Ravelo, A. C., D. H. Andreasen, M. Lyle, A. O. Lyle & M. W. Wara, 2004. Regional climate shifts caused by gradual global cooling in the Pliocene epoch. Nature 429: 263–267.
Ray, N. & J. M. Adams, 2001. A GIS-based vegetation map of the world at the Last Glacial Maximum (25,000–15,000 BP). Internet Archaeology 11 [available on internet at http://intarch.ac.uk/journal/issue11/bristow_index.html].
Ronquist, F. & J. P. Huelsenbeck, 2003. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572–1574.
Schmitt, T., 2007. Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Frontiers in Zoology 4: 11.
Schmitt, T., 2009. Biogeographical and evolutionary importance of the European high mountain systems. Frontiers in Zoology 6: 9.
Schmitt, T. & G. M. Hewitt, 2006. Molecular biogeography of the arctic-alpine disjunct burnet moth species Zygaena exulans (Zygaenidae, Lepidoptera) in the Pyrenees and Alps. Journal of Biogeography 31: 885–893.
Schmitt, T., G. M. Hewitt & P. Müller, 2006. Disjunct distributions during glacial and interglacial periods in mountain butterflies: Erebia epiphron as an example. Journal of Evolutionary Biology 19: 108–113.
Schmitt, T., C. Muster & P. Schönswetter, 2010. Are disjunct alpine and arctic-alpine animal and plant species in the western palearctic really “Relics of a cold past”? In Habel, J.C. & T. Assmann (eds), Relict Species Phylogeography and Conservation Biology. Springer, Berlin: 239–252.
Schönswetter, P., A. Tribsch, G. M. Schneeweiss & H. Niklfeld, 2003. Disjunctions in relict alpine plants: phylogeography of Androsace brevis and A. wulfeniana (Primulaceae). Botanical Journal of the Linnean Society 141: 437–446.
Senni, K., N. Fujii, H. Takahashi, T. Sugawara & M. Wakabayashi, 2005. Intraspecific chloroplast DNA variation of the alpine plants in Japan. Acta Phytotaxonomica et Geobotanica 56: 265–275.
Sheldon, T. A., N. E. Mandrak & N. R. Lovejoy, 2008. Biogeography of the deepwater sculpin (Myoxocephalus thompsonii), a Nearctic glacial relict. Canadian Journal of Zoology 86: 108–115.
Skrede, I., P. B. Eidesen, R. P. Portela & C. Brochmann, 2006. Refugia, differentiation and postglacial migration in arctic-alpine Eurasia, exemplified by the mountain avens (Dryas octopetala L.). Molecular Ecology 15: 1827–1840.
Sokolov, P. E., 2000. An improved method for DNA isolation from mucopolysaccharide-rich molluscan tissues. Journal of Molluscan Studies 66: 573–575.
Sonnenberg, R., A. W. Notle & D. Tautz, 2007. An evaluation of LSU rDNA D1-D2 sequences for their use in species identification. Frontiers in Zoology 4: 6.
Starobogatov, Ya. I., L. A. Prozorova, V. V. Bogatov & E. M. Sayenko, 2004. Molluscs. In Bogatov, V. V. & S. J. Tsalolikhin (eds), Key to Freshwater Invertebrates of Russia and adjacent lands, Vol. 6., Molluscus, Polychaetes, Nemerteans Nauka, St. Petersburg: 10–491.
Strong, E. E., O. Gargominy, W. F. Ponder & P. Bouchet, 2008. Global diversity of gastropods (Gastropoda; Mollusca) in freshwater. Hydrobiologia 595: 149–166.
Tanabe, A. S., 2011. Kakusan4 and Aminosan: two programs for comparing nonpartitioned, proportional and separate models for combined molecular phylogenetic analyses of multilocus sequence data. Molecular Ecology Resources 11: 914–921.
Tanabe, A. S., 2012. Phylogears version 2.2.2012.02.13 [available on internet at http://www.fifthdimension.jp/].
Väinölä, R., J. K. Vainio & J. U. Palo, 2001. Phylogeography of “glacial relict” Gammaracanthus (Crustacea, Amphipoda) from boreal lakes and the Caspian and White Seas. Canadian Journal of Fisheries and Aquatic Science 58: 2247–2257.
Varga, Z. S. & T. Schmitt, 2008. Types of oreal and oreotundral disjunctions in the western Palearctic. Biological Journal of the Linnean Society 93: 415–430.
Vinarski, M. & Y. Kantor, 2016. Analytical Catalogue of Fresh and Brackish Water Molluscs of Russia and Adjacent Countries. KMK Scientific Press, Moscow.
von Martens, E., 1877. Übersicht über die von Hilgendorf und Dönitz in Japan gesammelten Binnenmollusken. Sitzungsberichte der Gesellschaft natur- forschender Freunde zu Berlin 1877: 97–123.
Vos, R. A., 2003. Accelerated likelihood surface exploration: the likelihood ratchet. Systematic Biology 52: 368–373.
Wilke, T., R. Schultheiß & C. Albrecht, 2009. As time goes by: a simple fool’s guide to molecular clock approaches in invertebrates. American Malacological Bulletin 27: 25–45.
Xu, S., P. D. N. Hebert, A. A. Kotov & M. E. Cristescu, 2009. The noncosmopolitanism paradigm of freshwater zooplankton: insights from the global phylogeography of the predatory cladoceran Polyphemus pediculus (Linnaeus, 1761) (Crustacea, Onychopoda). Molecular Ecology 18: 5161–5179.
Yu, Y., A. J. Harris & X. J. He, 2010. S-DIVA (statistical dispersal–vicariance analysis): a tool for inferring biogeographic histories. Molecular Phylogenetics and Evolution 56: 848–850.
Yu, Y., A. J. Harris, C. Blair & X. He, 2015. RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Molecular Phylogenetics and Evolution 87: 46–49.
Acknowledgements
We are thankful to H. Fukuda for providing samples information. We also thank T. Shimada, J.U. Otani, Y. Murai, K. Kawabe, T. Haga, Omachi City Cultural Property Center, and Katata Fishermen’s Cooperative for collecting materials. Finally, we thank two anonymous referees for providing us with helpful comments on this manuscript. This study was funded in part by Japan Society for the Promotion of Science Research Fellow Grant Number 16J04692.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Diego Fontaneto
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Saito, T., Prozorova, L., Sitnikova, T. et al. Molecular phylogeny of glacial relict species: a case of freshwater Valvatidae molluscs (Mollusca: Gastropoda) in North and East Asia. Hydrobiologia 818, 105–118 (2018). https://doi.org/10.1007/s10750-018-3595-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-018-3595-y