
Abstract
Molecular characterization and genetic diversity analysis were performed using SSR molecular markers in a commercially important ornamental chrysanthemum (Dendranthema grandiflora Tzvelev) involving standard cultivars and newly evolved genotypes. The total number of alleles was found to be 113 with an average of 4.34 per locus using 26 polymorphic SSR primers out of the 35 total SSRs initially screened. An average of 90.53 percent polymorphism was observed in the characterized genotypes with an average number of 1.03 monomorphic and 3.31 polymorphic bands. The mean polymorphic information content, effective multiplex ratio (EMR), Shannon index (I), expected (He), observed heterozygosity (Ho), observed allele number (Na), effective allele number (Ne), marker index (MI), and resolving power (Rp) were observed at 0.63, 2.44, 1.00, 0.60, 0.69, 3.73, 2.60, 1.78 and 4.18, respectively. The minimum and maximum similarity values based on the Jaccard coefficient were observed to be 0.41 to 0.80. The population structure showed an admixture of three different genetic pools in the examined genotypes. The DARwin-based neighbor joining analysis also revealed two genotypes, namely UHFSCr-114 and UHFSCr-122, were more prominent than the rest of the chrysanthemum genotypes. Furthermore, it has already been observed in our previous studies that these two newly evolved genotypes were also found diversified in terms of their phenotypic characteristics. Therefore, in the present study, we observed high genetic variability among the studied genotypes as well as verified the mutant behaviour of newly evolved chrysanthemum genotypes at the molecular level. In addition, the current results would also accelerate and facilitate work on the release of highly diversified chrysanthemum genotypes as new cultivars in the near future.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chrysanthemum (Dendranthema grandiflora Tzvelev) is one of the widespread flower of significant aesthetic value. It is a segmental allohexaploid with a somatic chromosomal number of 54 (2n = 6x = 54) (Klie et al. 2014) and has a broad genome size of 9.4 Gb. It is grown widely all over the world and is an important economic ornament in many Southeast Asian and European countries, accounting for a large share of the flower industry (Zhang et al. 2011). Chrysanthemum is cultivated in the USA, Japan, Netherlands, Italy, Colombia, Spain, and Germany. There are 6,000 chrysanthemum plant cultivars listed by the National Chrysanthemum Society of Britain (Datta 2013). In India, the total development area for loose flowers and cut flowers is around 1991.4 and 867.1 ha, respectively. (Anonymous 2019). Chrysanthemum flowers have antibacterial, antifungal, and antiviral properties in addition to their aesthetic value. It also has anti-inflammatory medicinal properties that treat swelling owing to the stinging of the bee, broken muscles, and allergic reactions.
Chrysanthemum is one of the forms of perennials that begin to bloom early and is regarded as a favourite flower for the month of November. The height of the plant varies up to 1 m and in early winter, flowers bloom with a wide variety of colors, shapes and sizes. The distinctive family feature is that a large number of flowers are arranged on the flattened axis to produce a compact floral head that looks like a single flower. The flower has two types of florets, the inner one is called disk floret, containing both male and female reproductive parts while, the outer one is called ray floret with only female part and is unisexual. The wild taxa are characterized by their yellow disc florets. Chrysanthemum contains flavonoids such as luteolin, apigenin, acacetin, and choline. It's also shown to be a rich source of vitamin B1, vitamin C, vitamin A, niacin, folic acid, and pantothenic acid, as well as calcium, magnesium, iron, and phosphorus.
Plant genetic resources are one of the most valuable resources available to humans, so the characterization of germplasm is important to their useful and efficient management. A large number of cultivars with the necessary features are being cultivated due to the need to grow novel cultivars with quality characteristics. Many of these heirloom cultivars are being replaced by contemporary small genetic base cultivars as a result of multinational marketing and worldwide adoption, leading to a constant depletion of our traditional cultivars. Such characterization of germplasm is essential not only for identifying different species and providing information on accessions that ensures the most effective use of germplasm collections but also for determining genetic connectivity among them and addressing Intellectual Property Rights (IPR) issues, as well as their use and conservation in any crop improvement programme.
Consequently, being an economically important cultivated flower crop, chrysanthemum genetic diversity estimation becomes a critical component of effective breeding and development of new cultivars. The process of developing new varieties involves several steps and takes a year, particularly in the case of horticultural fruits and plants. As may be observed in the numerous varieties, cultivated chrysanthemums exhibit a lot of genetic variation. Traditional breeding programmes may not be adequate to develop complex traits in chrysanthemums due to the accumulated genetic variability, which provides a significant resource of alleles for improving important decorative traits. Identification of varieties and breeding lines becomes essential in crops like chrysanthemum, where the origin of varieties is often unknown.
Assessment of diversity based on morphological parameters has always been restricted by the fact that they have large effects on phenotype, masking the influence of a related minor gene, making it difficult to identify desirable linkage for choice and their weakness of being extremely affected by the environment; therefore, they may not be suitable for detailed research. However, isozyme assessment has inherent drawbacks, such as a limited number of suitable enzymes loci in genome expression and enzymes dependent on growth and seasonal development. Consequently, DNA-based markers have been evolved as a reliable tool in germplasm identification, characterization, diversity analysis, detection of redundancies in gene bank collections, observing genetic changes during germplasm preservation, and gene tagging after the advent of molecular biology techniques. In genetic diversity/similarity assessments, DNA has been characterized as an optical molecule because of certain desirable properties, such as plasticity, ubiquity and stability (Caetano-Anolles et al. 1991).
As a result, molecular markers outperform morphological and biochemical identifiers when they are being used in interpreting genetic variation and interactions. Owing to their DNA level polymorphism identification aspect based on PCR (Polymerase Chain Reaction), the genetic makers gained the respective popularity. Additionally, due to their abundance, DNA markers became the most commonly used marker type. In comparison to morphological and biochemical markers, the relevant environmental factors and growth phases do not affect DNA markers. Moreover, molecular markers have been proved as reliable tools in the development of genetic linkage maps and marker-assisted selection (MAS) breeding (Negi et al. 2020a). Among the various molecular markers, SSR markers because of their co-dominant inheritance, high rate of polymorphism, abundance in eukaryotic genomes, reproducibility and relative ease of analysis have become a marker of choice for establishing unique genetic identities or fingerprints, exploring genetic relatedness between accessions, and assessing genetic diversity within a collection (Schlotter 2004). Also, the SSR markers have been successfully employed by various researchers to study genetic relationships, defining elite alleles through interaction and association in chrysanthemum cultivars (Khaing et al. 2013; Li et al. 2013; Wang et al. 2013; Zhang et al. 2014; Min-Jo et al. 2015; Liu et al. 2015; Park et al. 2015; Feng et al. 2016; Chang et al. 2018; Han et al. 2018).
Therefore, the present study aimed to find out the level of genetic diversity by characterizing the newly evolved genotypes with the standard cultivars of chrysanthemum at the molecular level by using SSR markers. The newly evolved genotypes have already been characterized by our group on phenotypic attributes viz., growth, flowering behavior, performance, and stability analysis in two different locations at the experimental farm of Department of Floriculture and Landscape Architecture, Dr Yashwant Singh Parmar University of Horticulture and Forestry, Nauni, Solan (HP) India and ICAR- Indian Agricultural Research Institute Regional Research Station, Katrain, Kullu (HP) India including, cultivar ‘Ajay’ for cut flower production (Negi et al. 2018, 2020b, 2020c) and ‘Surf’ as loose flower production (Negi et al. 2019) as standard checks, respectively.
Materials and methods
Molecular characterization
A total 36 chrysanthemum genotypes including 15 standard cultivars and 21 newly evolved genotypes were considered for the molecular characterization (Table 1, Suppl Figs. 1, 2, 3). The CTAB method (Doyle and Doyle 1990) was used to isolate genomic DNA from young and fresh leaves of the chrysanthemum genotypes. Standard spectrophotometry was used to assess the quantity and purity of the DNA (Ausubel et al. 1995) using a nanodrop spectrophotometer (Eppendorf Biophotometer, Germany). A total of 35 highly polymorphic SSR primer pairs were chosen based on their uniform distribution across the chrysanthemum genome for performing molecular characterization studies (Wang et al. 2013; Feng et al. 2016) (Suppl Table 1). A final volume of 15 µl reaction mixture containing 1X Taq polymerase buffer with MgCl2 (Genei, India), 0.2 mM dNTP (Genei, India), 0.3U Taq DNA polymerase (Genei, India), 10 pmol SSR primers (Eurofins, India) and 50 ng/µl of genomic DNA was used in PCR based studies. The amplification was carried out in a thermal cycler (Applied Biosystem, USA) with an initial denaturation step of 94 °C for 4–5 min, followed by 35 cycles of 94 °C for 1 min, Primer Ta for 1 min, and 72 °C for 1 min, with a final extension step of 72 °C for 8–10 min. The amplified DNA was thoroughly mixed with 6X loading dye before being separated on a 3.0% agarose gel using 50 and 100 bp ladders to estimate band size, and the gels were documented by using Gel Documentation System (Syngene, UK).
Genetic diversity analysis
The data of DNA banding profiles obtained on electrophoresed agarose gels based on the binary matrix was subsequently processed using analytical softwares viz., NTSYS-PC ver. 2.02i and DARwin ver.6 for analyzing similarity coefficients and tree-formed dendrogram construction. The polymorphic information content (PIC) exhibited by each primer, Marker Index (MI), effective multiplex ratio (EMR), Resolving Power (Rp) along with the estimates of gene diversity for each population across all loci in terms of alleles per locus (Na), effective number of allele (Ne), Shannon’s information index (I), observed heterozygosity (Ho) and expected heterozygosity (He) were calculated using program POPGENE 1.32 (Nei 1978; Yeh et al. 1999). To understand the genetic structure among the characterized genotypes, investigation of the population using multi-locus genotypic data by estimating the frequencies of population allele in situations where many individuals migrated or admixed was analyzed by using the software STRUCTURE version 2.3.4 (Pritchard et al. 2000).
Results
Molecular characterization
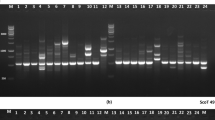
Out of the total of 35 SSR primers, only 26 were found to be polymorphic and further used to amplify the genomic DNA of chrysanthemum genotypes for clear amplified banding profiles under optimal conditions. In the banding pattern across 36 chrysanthemum genotypes, a varying amount of polymorphism was identified. There were 113 amplified bands produced by the total 26 SSR primer pairs, with an average of 4.34 amplified bands per primer, of which 86 (76.10%) were polymorphic and 27 (23.90%) were monomorphic. Consequently, 3.31 polymorphic and 1.03 monomorphic bands were found on an average per primer (Table 2). The means; 3.73 number of alleles (Na), 2.60 effective number of alleles (Ne), 1.00 Shannon index (I) were obtained, while expected heterozygosity (He) and observed heterozygosity (Ho) were observed with mean values of 0.60 and 0.69, respectively. Maximum number of alleles produced ranged from 2 to 8 with CMeSSR014 primer. The mean allele frequency was 2.10, ranging from 1.05 to 3.58, with the maximum observed in SSR primer 313, while minimum in SSR primer 320. Amplified products ranged in size from 50 to 400 bp. SSR primer 984 exhibited the least diversity among all the 26 polymorphic primers considered in the molecular characterization study (Fig. 1). The highest polymorphism of 87.5% was recorded for SSR primer CMeSSR014, while the lowest 50% polymorphism was observed among six SSR primers namely; 320, 1484, 984, CMeSSR018, gi298296818, and 1584 with an average of 90.53 percent. The mean values; 0.63 polymorphic information contents (PIC), 2.44 effective multiplex ratio (EMR), 1.78 marker index (MI) and 4.18 resolving power (Rp) values were were obtained in the inferred diversity.

DNA profiles obtained using SSR primer 984 G1 to G36: chrysanthemum genotypes, L1-50 bp ladder, L-100 bp ladder
Genetic diversity analysis
The chrysanthemum genotypes were divided into two major clusters, with cluster A including only three genotypes and cluster B containing 31 genotypes, which bifurcated at a similarity coefficient value of 0.58 based on the Jaccard coefficient using NTSYS-pc program (Fig. 2). The genotypes ‘UHFSChr-122’ and ‘UHFSChr-114’ were found to be highly diversified and did not show any grouping with other chrysanthemum genotypes. Cluster A comprised three genotypes namely; UHFSChr-125, UHFSChr-113 and UHFSChr-131 merged at a similarity coefficient of 0.64. At a similarity value of 0.60, the major cluster B was further subdivided into two groups B1 and B2. There were 28 genotypes in sub-cluster B1, which were further divided into two groups comprising 21 and 7 genotypes, respectively. Similarly, in sub-clusters B2, the genotype ‘William Turner’ was found independent whereas, the ‘UHFSChr-130’ and ‘UHFSChr-56’ genotypes were grouped in the same cluster. The similarity coefficient of Jaccard varied from 0.41 to 0.80, with the highest similarity between genotypes 'UHFSChr-111' and 'Thaiching Queen' and the lowest similarity between ‘UHFSChr-114’ and ‘UHFSChr-122’. The factorial analysis was done by using DARWin ver.6 software in which the genotype named ‘UHFSChr-113’, ‘UHFSChr-125’ and ‘UHFSChr-131’ were found to be grouped together. The genotype ‘UHFSChr-114’ was found to be distinct from the rest of the other genotypes (Fig. 3). Equally, the neighbor-joining cluster analysis with bootstrap values using DARwin software also confirmed the distinctiveness of these two genotypes viz., ‘UHFSChr-114’ and ‘UHFSChr-122’ (Fig. 4) on the same line that was interpreted from the dendrogram obtained using NTSYS software.

Dendrogram obtained from SSR analysis showing genetic relationships among chrysanthemum genotypes using NTSYS pc ver.2.02i

Factorial analysis using SSR marks in chrysanthemum genotypes

Diversity interpretation among chrysanthemum genotypes using SSR analysis
To determine the population structure of 36 chrysanthemum genotypes considered in the present study, the Bayesian-based cluster analysis was performed using STRUCTURE version 2.3.4. The analysis was performed using admixture model assumptions with correlated alleles; K was presumed to be 1–10, selected after five independent runs. STRUCTURE HARVESTER software (Earl and von Holdt 2012) was used to collate the results obtained from STRUCTURE software and the maximum value of ΔK for SSR marker data was observed for K = 3. The Evanno’s test also found a maximum peak value for Delta K at K = 3 in the plots of L (K) versus Delta confirming a likely assignment of the chrysanthemum germplasm into three sub-groups. The slope at K = 3 was found to be sharper and steeper than other values (Fig. 5). The 36 genotypes of chrysanthemum showed an admixture of three different genetic pools (Fig. 6).

Kinship value (K = 3) obtained among chrysanthemum genotypes

Population structure interpretation based on SSR profiling in chrysanthemum genotypes
Discussion
The molecular characterization, genetic diversity analysis and population structure analysis is one of the essential pre-requisites in any crop improvement programme. In the present study, we have accessed molecular characterization of 36 genotypes of chrysanthemum containing cultivars and newly evolved genotypes by using 26 polymorphic SSR primer pairs. The number of amplified bands produced was found to be in the range of 2–8, while the size of amplified products ranged from 50–400 bp. The maximum and minimum polymorphism was observed 87.50% and 50.00%, respectively (Table 2). Correspondingly, maximum average percent polymorphism of 97.14 and 86.21 with amplicon size 135–500 bp and 80–400 bp was reported by Khaing et al. (2013) and Chang et al. (2018), respectively while, minimum polymorphism percentage of 31.55 with amplicon size 120–1070 bp was observed by Samarina et al. (2021) among chrysanthemum cultivars.
In the present study, PIC, EMR, I, He, Ho, Na, Ne, MI, and Rp values ranged from 0.35 to 0.81, 0.5 to 6.12, 0.35 to 1.54, 0.22 to 0.81, 0.25 to 1.00, 2 to 6, 1.28 to 4.43, 0.17 to 4.41, and 2.11 to 7.16, respectively (Table 2). Compared with the previous studies on chrysanthemum, Khaing et al. (2013) found PIC, Na and Heterozygosity value in the range of 0.22 to 0.88, 4 to 27 and 0.18 to 0.92, respectively, whereas Zhang and Wang (2013) observed Na and heterozygosity in the range of 2 to 14 and 0.05 to 0.89. Likewise, Zhang et al. (2014) found Na, Ne, I, H values in the range 2 to 22, 1.78 to 9.28, 0.23 to 0.65, 0.26 to 0.67, respectively. Min-Jo et al. (2015) observed the polymorphic information content and heterozygosity values range of 0–0.88 and 0–0.89 while, Yuan et al. (2016) observed Na, Ne, He, Ho and I values in the range of 8–19, 1.89–6.24, 0.48–0.87, 0.30–0.80 and 1.15–2.44, respectively, by using 16 newly developed SSR in chrysanthemum. Likewise, Chang et al. (2018) observed Na, Ne, and I values ranging from 2 to 6, 1.34 to 4.74, and 0.39 to 0.66, while Kobeissi et al. (2018) observed PIC ranging from 0 (CMeSSR003, JH1, JH15, and KNUCRY-59) to 0.79 (CMeSSR001) with Na, Ne, I, Ho and He values ranging from 1–4, 1–2.98, 0–1.09, 0–0.96 and 0–0.54, respectively. Also, Mekapogu et al. (2020) observed an average polymorphism of 0.41 with PIC value of 7 SSRs ranging from 0.25 to 0.60. Samarina et al. (2021) observed PIC value range of 0.38 to 0.50 and Shannon index value range of 0.31 to 0.63. SSRs in this sample were mainly 12 to 20 bp long (93.7%), suggesting a modest level of polymorphism. A huge amount of diversity was inferred by various researchers in their earlier studies that might be due to the number of genotypes characterized, number of polymorphic SSR primers used and the geographic origin of the genotypes for varying polymorphism ratios. Similarly, Liu et al. (2015) also suggested that the number of polymorphic SSRs could differ if more primer pairs were initially screened, which would provide greater validity to the predicted genetic diversity.
In this study, Jaccard’s similarity coefficients were obtained in the range of 0.41–0.80 that were found similar to the previous studies conducted in chrysanthemum by Chang et al. (2018) i.e., 0.53–0.88 and by Kumari et al. (2017) in sunflower i.e., 0.20–0.90. The obtained dendrogram grouped the chrysanthemum genotypes into two major clusters where the genotypes ‘UHFS Chr-114’ and ‘UHFS Chr-122’ were reported to be most diverse (Fig. 2). These results were also in agreement with the previous studies conducted in chrysanthemum cultivars and accessions (Feng et al., (2016); Chang et al., (2018). In contrast to the present findings, Olejnik et al. (2021) clustered 97 chrysanthemum cultivars into four major groups by using 14 SSR markers. The factorial analysis done using DARWin software showed the genotype ‘UHFS Chr-114’ highly diverse from the rest of the other genotypes, which is in agreement with the results obtained in cluster analysis (Fig. 3). Similar to the present study, Yang et al. (2006), Zhang et al. (2014), Feng et al. (2016) and Chang et al. (2018) also revealed that the results of Principal Component Analysis (PCA) exhibited similarity with the clustering analysis.
In the present study, the population structure showed an admixture of three different genetic pools among 36 chrysanthemum genotypes (Figs. 5&6), and were agreement in a similar line with the previous study performed in ornamental crops like chrysanthemum (Khaing et al. 2013) and rose (Agarwal et al. 2019). Whereas, Olejnik et al. (2021) and Samarina et al. (2021) observed the chrysanthemum cultivars grouping into four and two admixtures with peak values of K = 4 and K = 2, respectively. Moreover, domestication history, breeding, resource exchange, high heterozygosity, and chrysanthemum self-incompatibility may all contribute to these mixed populations (Anderson 2006; Zhang et al. 2010; Zhao et al. 2010; Roein et al. 2014). Therefore, being a cross-pollinated crop and polyploidy level of the genome, chrysanthemum showed a high level of genetic variability among the studied genotypes.
The identified highly diversified newly evolved genotypes on the basis of molecular characterization studies were further compared and analyzed based on their already studied phenotypic attributes viz., growth, flowering behavior, performance, and stability using standard checks namely, ‘Ajay’ for cut flower production (Negi et al. 2018, 2020b, 2020c) and ‘Surf’ as loose flower production (Negi et al. 2019). The revealed molecular observations were found in the same line with our previous finding based on phenotypic attributes conducted by Negi et al. (2018), where amongst the total 18 newly evolved genotypes, the genotype ‘UHFS Chr-122’ was found to be diversified with a minimum number of days for flowering. Also, the genotype ‘UHF Chr-114’ was observed with a maximum height and the lowest stem weight (Negi et al. 2020b).
From the present study, it was concluded that SSR markers have been found to be an efficient tool for deciphering a high level of genetic diversity among the genotypes studied. The resulting molecular data confirm the divergence in two newly evolved chrysanthemum genotypes. These studies would assist in the development of trait-specific markers and provide a platform to confirm the actual mutant characters of these newly developed chrysanthemum genotypes. Additionally, these findings would expedite and facilitate work on the release of highly diverse chrysanthemum genotypes as a new cultivar in the near future.
References
Agarwal A, Gupta V, Ul Haq S, Jatav PK, Kothari SL, Kachhwaha S (2019) Assessment of genetic diversity in 29 rose germplasms using SCoT marker. J King Saud Univ Sci 31:780–788. https://doi.org/10.1016/j.jksus.2018.04.022
Anderson NO (2006) Chrysanthemum Dendranthema grandiflora Tzvelv. In: Flower breeding and genetics: issues, challenges, and opportunities for the 21st century. In: Anderson NO (eds). Springer, Dordrecht, pp 389–437
Anonymous (2019) Area and production of horticulture crops for 2018–2019. http://nhb.gov.in/Statistics/Default.aspx
Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (1995) Short protocols in molecular biology. Wiley, New York, p 836
Caetano-Anolles G, Bassam BJ, Gresshoff PM (1991) DNA fingerprinting using very short arbitrary oligonucleotide primers. Biotechnology 9:553–557. https://doi.org/10.1038/nbt0691-553
Chang L, Dongliang C, Cheng X, Hua L, Yahui LI, Hunag C (2018) SSR analysis of genetic relationship and classification in chrysanthemum germplasm collection. Hortic Plant J 4:73–82. https://doi.org/10.1016/j.hpj.2018.01.003
Datta SKD (2013) Chrysanthemum morifolium (Ramat) a unique genetic material for breeding. Sci Cult 79:307–313
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15. https://doi.org/10.1007/978-3-642-83962-7_18
Earl D, Von Holdt B (2012) Structure Harvester: a website and program for visualizing structure output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Feng S, Renfeng H, Jiangjie L, Jiang M, Shen X, Jiang Y, Wang Z, Wang H (2016) Development of SSR markers and assessment of genetic diversity in medicinal Chrysanthemum morifolium cultivars. Front Genet 7:13. https://doi.org/10.3389/fgene.2016.00113
Han Z, Ma X, Wei M, Zhao ZR, Chen W (2018) SSR marker development and intraspecific genetic divergence exploration of Chrysanthemum indicum based on transcriptome analysis. BMC Genom 19:1–10. https://doi.org/10.1186/s12864-018-4702-1
Khaing AA, Moe KT, Hong WJ, Park CS, Yeon KH, Park HS, Kim DC, Choi BJ, Jung YJ, Chae SC, Lee KM, Park YJ (2013) Phylogenetic relationships of chrysanthemums in Korea based on novel SSR markers. Genet Mol Res 12:5335–5347. https://doi.org/10.4238/2013.November.7.8
Klie M, Schie S, Linde M, Debener T (2014) The type of ploidy of chrysanthemum is not black or white: a comparison of a molecular approach to published cytological methods. Front Plant Sci 5:479. https://doi.org/10.3389/fpls.2014.00479
Kobeissi B, Saidi A, Kobeissi A, Shafie M (2018) Applicability of SCoT and SSR molecular markers for genetic diversity analysis in Chrysanthemum morifolium genotypes. Proceed National Acad Sci India Sect b Biol Sci 89:1067–1077. https://doi.org/10.1007/s40011-018-1024-7
Kumari S, Choudhary RM, Kumara SRV, Saharan V, Joshi A, Munot M (2017) Assessment of genetic diversity in safflower (Carthamus tinctorius L.) genotypes through morphological and SSR marker. J Pharmacogn Phytochem 6:2723–2731
Li YH, Luo C, Wu ZY, Zhang XH, Cheng X, Dong R, Huang CL (2013) Microsatellite enrichment by magnetic beads in chrysanthemum. Acta Hort 977:269–278. https://doi.org/10.17660/ActaHortic.2013.977.32
Liu H, Zhang QX, Sun M, Pan HT, Kong ZX (2015) Development of expressed sequence tag-simple sequence repeat markers for Chrysanthemum morifolium and closely related species. Genet Mol Res 14:7578–7586. https://doi.org/10.4238/2015.July.13.1
Mekapoku M, Kwon OK, Hyun DY, Lee KJ, Ahn MS, Park JT, Jung JA (2020) Identification of standard type cultivars in chrysanthemum (Dendranthema grandiforum) using SSR markers. Hortic Environ Biotechnol 61:153–161. https://doi.org/10.1007/s13580-019-00201-0
Min-Jo K, Chu H, Lian S, Cho WK (2015) Development of EST-derived SSR markers using next-generation sequencing to reveal the genetic diversity of 50 chrysanthemum cultivars. Biochem Syst Ecol 60:37–45. https://doi.org/10.1016/j.bse.2015.03.002
Negi R, Dhiman SR, Gupta YC (2018) Studies on growth and flowering behavior of newly evolved genotypes of chrysanthemum (Dendranthema grandifloraT zvelev) for cut flower production. Int J Sci Res 8:745–747
Negi R, Dhiman SR, Gupta YC (2019) Studies on growth and flowering behavior of newly evolved genotypes of chrysanthemum (Dendranthema grandiflora Tzvelev) for loose flower production. Int J Curr Microbiol App Sci 8:341–346. https://doi.org/10.20546/ijcmas.2019.811.043
Negi S, Sharma G, Sharma R (2020a) Introgression and confirmation of everbearing trait in strawberry (Fragaria x ananassa Duch.). Physiol Mol Biol Plants 26:2407–2416. https://doi.org/10.1007/s12298-020-00916-w
Negi R, Dhiman SR, Dhiman MR, Gupta YC, Dogra RK, Gupta RK (2020b) Performance and flower characterization of newly evolved genotypes of chrysanthemum (Dendranthema grandiflora Tzvelev) for cut flower production. Int J Chem Stud 8:2921–2923. https://doi.org/10.22271/chemi.2020.v8.i4ai.10093
Negi R, Dogra RK, Dhiman SR, Gupta YC, Gupta RK, Dhiman MR (2020c) Stability analysis of newly evolved genotypes of chrysanthemum (Dendranthema grandiflora Tzvelev) for cut flower production using Eberhart and Russel model. Int J Curr Microbiol Appl Sci 9:477–486. https://doi.org/10.20546/ijcmas.2020c.906.062
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590. https://doi.org/10.1093/genetics/89.3.583
Olejnik A, Parkitna K, Kozak B, Florczak S, Matkowski J, Nowosad K (2021) Assessment of the genetic diversity of chrysanthemum cultivars using SSR markers. Agron 11:2318. https://doi.org/10.3390/agronomy11112318
Park SK, Arens P, Esselink D, Lim JH, Shin HK (2015) Analysis of inheritance mode in chrysanthemum using EST-derived SSR markers. Scientia Hort 192:80–88. https://doi.org/10.1016/j.scienta.2015.05.009
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
Roein Z, Asil MH, Sabouri A, Dadras AR (2014) Genetic structure of chrysanthemum genotypes from Iran assessed by AFLP markers and phenotypic traits. Plant Syst Evol 300:493–503. https://doi.org/10.1007/s00606-013-0898-3
Samarina LS, Malyarovskaya VI, Reim S, Yakushina LG, Koninskaya NG, Klemeshova KV, Shkhalakhova RM, Matskiv AO, Shurkina ES, Gabueva TY, Slepchenko NA (2021) Transferability of ISSR, SCoT and SSR markers for Chrysanthemum× Morifolium Ramat and genetic relationships among commercial Russian cultivars. Plants 10:1302. https://doi.org/10.3390/plants10071302
Schlotter C (2004) The evolution of molecular markers- a matter of fashion. Nat Rev Genet 5:63–69. https://doi.org/10.1038/nrg1249
Wang H, Jiang J, Chen S, Qi X, Peng H, Li P, Song A, Guan Z, Fang W, Liao Y, Chen F (2013) Next-generation sequencing of the Chrysanthemum nankingense (Asteraceae) transcriptome permits large-scale unigene assembly and SSR marker discovery. PLoS ONE 8:e62293. https://doi.org/10.1371/journal.pone.0062293
Yang W, Glover BJ, Rao GY, Yang J (2006) Molecular evidence for multiple polyploidization and lineage recombination in the Chrysanthemum indicum polyploid complex (Asteraceae). New Phytol 171:875–886. https://doi.org/10.1111/j.1469-8137.2006.01779.x
Yeh FC, Yang RC, Boyle T, Ye ZH, Mao JX (1999) Popgene. Microsoft windows-based freeware for population genetic analysis. Release 1:31. http://www.ualberta.ca/~fyeh/,1999-ci.nii.ac.jp
Yuan WJ, Ye S, Du LH, Li SM, Miao X, Shang F (2016) Isolation and characterization of microsatellite markers for Dendranthema morifolium (Asteraceae) using next- generation sequencing. Genet Mol Res 15:1–7. https://doi.org/10.4238/gmr.15048765
Zhang F, Chen S, Chen F, Fang W, Li F (2010) A preliminary genetic linkage map of chrysanthemum (Chrysanthemum morifolium) cultivars using RAPD, ISSR and AFLP markers. Scientia Hort 125:422–428. https://doi.org/10.1016/j.scienta.2010.03.028
Zhang F, Chen S, Chen F, Fang W, Chen Y, Li F (2011) SRAP-based mapping and QTL detection for inflorescence-related traits in chrysanthemum (Dendranthema morifolium). Mol Breed 27:11–23. https://doi.org/10.1007/s11032-010-9409-1
Zhang Y, Dai S, Hong Y, Song X (2014) Application of genomic SSR locus polymorphisms on the identification and classification of chrysanthemum cultivars in China. PLoS ONE 9:e104856. https://doi.org/10.1371/journal.pone.0104856
Zhang Y, Wang C (2013) Assessing the genetic diversity of chrysanthemum cultivars with microsatellites. J Amer Soc Hort Sci 138:479–486. https://doi.org/10.21273/JASHS.138.6.479
Zhao WG, Chung JW, Cho YI, Rha WH, Lee GA, Ma KH, Han SH, Bang KH, Park CB, Kim SM, Park YJ (2010) Molecular genetic diversity and population structure in Lycium accessions using SSR markers. CR Biologies 333:793–800. https://doi.org/10.1016/j.crvi.2010.10.002
Acknowledgements
The authors are grateful to the Indian Council of Agricultural Research (ICAR), New Delhi, India for providing funds through a central assistance scheme.
Author information
Authors and Affiliations
Contributions
AT: conducted PCR-based studies, data recording, writing rough draft of the manuscript; RS: conceptualization, methodology, overall supervision, manuscript editing and finalization; SRD: procurement of genotypes, data recording on phenotypic attributes, manuscript editing; RN: data recording on phenotypic attributes, data analysis; AS: molecular data recording. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.



Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Thakur, A., Sharma, R., Dhiman, S.R. et al. Genetic diversity analysis in chrysanthemum (Dendranthema grandiflora Tzvelev) using SSR markers: corroborating mutant behaviour of newly evolved genotypes. Genet Resour Crop Evol 70, 449–460 (2023). https://doi.org/10.1007/s10722-022-01438-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-022-01438-y