
Abstract
Algerian maize has been cultivated in Saharan Oases for many centuries, determining its adaption to extreme environments. Therefore, maize landraces from Sahara could be considered as valuable genetic resources for breeding. Morphological and molecular characterization of fifty-six populations were assessed using 14 agro-morphological traits and 18 SSR markers. Populations were evaluated in field experiment in an augmented randomized complete block design. ANOVA on morphological data revealed significant difference among populations. Analysis of principal component showed two principal components describing 55.44% of total variation. Flowering time, plant height, ears traits and yield were the most discriminatory traits. Genetic analysis identified a large number of alleles (191) with mean value of 10.61 alleles per locus. High average PIC value (0.57) indicates informativeness of the selected markers in this study. The genetic structure analysis revealed a high genetic differentiation (Fst = 0.22) among populations, showing a greater genetic diversity within Algerian populations than among them. Bayesian model-based structure analysis assigned genotypes into two groups. Both phenotypic and SSR analysis revealed significant genetic diversity; albeit a clustering based on geographic origin was not observed. The wide genetic diversity of Saharan maize populations could be used as genetic resources in future maize breeding programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Maize (Zea mays L.) is considered the major staple food in the world and the most cultivated among cereals (Shiferaw et al. 2011), therefore, due to climate change about 20% of maize production is lost each year (Chen et al. 2012). Thus, maize improvement for abiotic stress tolerance could become a decisive goal and the future challenge for breeding programs (Betrán et al. 2003). To date, only 10% of total maize diversity is used in breeding programs worldwide (Beyenne et al. 2006a), and this narrow genetic background could be scarce to select for abiotic stress tolerances.
Morphological, biochemical and molecular characterization are now available for studying genetic diversity (Govindaraj et al. 2015). Although morphological and biochemical characterization were largely used in maize (Franco et al. 2001), both methods are highly affected by environment (Smith and Smith 1992; Beyenne et al. 2006a). By contrast, the DNA-based molecular marker techniques, such as simple sequence repeat (SSR), appeared more useful because not influenced by environmental changes and able to provide a direct measure of genetic diversity avoiding the genotype-environment interaction (Cömertpay et al. 2012; Govindaraj et al. 2015). SSR were largely used for analyzing maize genetic diversity (Smith and Smith 1992; Messmer et al. 1993; Warburton et al. 2002; Dubreuil et al. 2006) and their success is mainly due to high level of polymorphism, codominance, repeatability and reliability (Legesse et al. 2007). However, the genetic characterization of open-pollinated maize varieties is very expensive and time consuming (Prasanna 2012), and to overcome these limitations, population bulk DNA fingerprinting method was employed to analyze few bulked samples rather than many individual plants (Eschholz et al. 2010).
Many studies on African maize genetic diversity were already carried out (Beyene et al. 2006a; Legesse et al. 2007; Adeymo et al. 2011), but not many reported landraces from Algeria (Djemel et al. 2012; Aci et al. 2013, 2018).
After its introduction in Algeria by Arabs and Moors during the five centuries (McCann 2005), maize was grown especially in Saharan Oases, where was exposed to high temperature and drought stress compared to its traditional area of cultivation, and the large genetic diversity available in maize open pollinated populations resulted in the adaptation to this extreme environmental condition.
Commonly, F1 hybrid have not been available to small-scale farmers localized in the maize zone diversity (Adrar province) described by Djemel et al. (2012), where the geographical isolation from maize hybrid-land in Ghardaia province (Algeria) was guaranteed.
Further, the F1 genetic background is generally narrow and less adaptable to the drastic climate of the Algerian desert. Thus, the cultivation of open pollinated varieties (OPVs) appears most appropriate for the purpose small surfaces cultivation in which the OPVs are cultivated year after year utilizing the selected seeds from the previous season to feed human and animal populations. As the Sahara Oases for maize cultivation are significantly distant one from each other, farmers maintained their own landraces, leading to high genetic and phenotypic variability mainly important for breeding programs (Hoxha et al. 2004; Wietholter et al. 2008). To avoid the influence of the new maize-growth systems on both crop diversity maintenance and traditional seed-saving, our most scientific priorities is to collect, preserve, and exploit the maize biodiversity from the Algerian Oases before its genetic erosion or contamination.
The first Algerian maize characterization, based on agro-morphological traits, identified a wide range of variability for adaptation to high temperature (Djemel et al. 2012), and these observations were confirmed by Aci et al. (2013, 2018), assessing the genetic diversity of maize populations using SSR markers.
In the present study, fifty-six Algerian maize populations collected from Saharan Oases were characterized using agro-morphological traits as well as SSR markers. For a first instance, this study permitted to increase the knowledge of maize genetic diversity from Algeria useful to establish a core collection to be used in future breeding programs.
Materials and methods
Plant material
Fifty-six maize open-pollinated populations, well-maintained at the National School of Agronomy of Algiers (ENSA), were selected as representative of the Oases Saharan maize. The populations were mainly collected from Adrar and Tamanrasset in 2009 and 2010 (Suppl. Table 1).
Field experiment
The maize populations were field assayed in 2013 in an augmented block design (Federer 1956) with three blocks and seven checks, of which two synthetic or landrace from USA (BS17, BSL), one from Canada (Longfellow) and four populations from Spain (Norteno, Rastrojero, Tuy and Tremesino), at the High National School of Agronomy (36° 47′ N, 2° 03′ E) located in the North of Algeria. Each plot consisted on one row 6 m length. The rows were spaced 0.70 m apart, and the hills were spaced 0.20 m apart to obtain a final density of 6 plants m−2. For each population, data of 14 morphological traits were estimated using 10 randomly selected plants per population based on CIMMYT and IBPGR (1991) maize descriptor as reported in Suppl. Table 2.
Phenotypic data analysis
Means for each morphological trait were analyzed using augmented complete block design adjusting by blocking effects of the replicated checks using ACBD-R software by CYMMYT (International Maize and Wheat Improvement Center), and the data were further analyzed to highlight significant differences by analysis of variance (ANOVA). Further, principal component analysis (PCA) was performed to identify which trait efficiently differentiated maize populations by using R software (R Development Core Team 2008). Before carrying out PCA, the mean of each trait was standardized for the deletion of scaling difference.
DNA extraction and PCR analysis
DNA was extracted from each population using bulk DNA fingerprinting method described by Rebourg et al. (2001). Each bulk was prepared by pooling an equal amount of leaf material from 15 individuals per population. After pooling and grounding steps from 100 mg of leaf material, using liquid nitrogen, DNA extraction was performed using DNeasy Plant Mini Kit (Qiagen, Milano, Italy), according to manufacturer protocol. The fifty-six populations were genotyped with a set of 18 SSR markers distributed throughout the genome (Aci et al. 2013, 2018). PCR reaction was carried out in 20 μL volume containing 20 ng DNA for each bulk, 1 U Taq DNA polymerase (Thermo Fisher Scientific Inc.), 0.32 μM reverse primer, 0.16 μM forward and 0.16 μM fluorescence (FAM) labeled universal primer M13 (− 21) as previously reported (Schuelke 2000; Carimi et al. 2011). Thermal cycling consisted of an initial denaturation step at 95 °C for 5 min, 30 cycles at 94°C/30 s, 56 °C/45 s and 72 °C/45 s followed by 8 cycles at 94 °C/30 s, 53 °C/45 s and 72 °C/45 s. The final step was the extension at 72 °C for 10 min. SSR products were separated in capillary sequencer ABI 3500 (Applied Biosystems). SSR allele size was analyzed using Gene Mapper v.5.0 software (Applied Biosystems). In particular, to estimate the allele frequencies for each bulk filtering allele calling “FreqsR” software was used. The allele sizes of each individual starting from bulk allele frequencies were calculated using “F-to-L” software (Warburton et al. 2002; Dubreuil et al. 2006), the analysis was performed by R platform (R Development Core Team 2008).
Genetic diversity analysis
To determine genetic parameters such as number of alleles (N), number of different alleles (Na), number of effective allele (Ne), expected (He) and observed (Ho) heterozygosity and Shannon diversity index (I) among populations and SSR loci GenAlex software version 6.3 (Peakall and Smouse 2006) was used. Private allele and polymorphic index content (PIC) for each SSR locus were determined using GDA (https://phylogeny.uconn.edu/software) and Cervus v.3.0.7 software (Copyright Tristan Marshal, Field Genetic, Ltd), respectively. Analysis of molecular variance (AMOVA) was also performed to analyze genetic variation among and within individuals (Excoffier et al. 1992) by Arlequin software (http://cmpg.unibe.ch/sofware/arlequin3), testing Fst by 9999 random permutations. Furthermore, a dendrogram was depicted based on Nei and Li (1979) pairwise distances matrix (SSR data-based) and the algorithm UPGMA (unweighted pair group method of arithmetic clustering) (Sneath and Sokal 1973) by using MEGA v. 6 software (Kumar et al. 2015). Moreover, a principal coordinate analysis (PCoA) was performed using GenAlex software version 6.3 (Peakall and Smouse 2006).
Finally, a model-based (Bayesian) clustering was performed to evaluate genetic relationship among individuals and population structure by using STRUCTURE software (Pritchard et al. 2000). The program was set up and run as reported in Mercati et al. (2013). Then, the criterion (ΔK) by Evanno et al. (2005) was used to determine the most probable K value, counteracting for the overestimation of subgroup number by STRUCTURE: lines with membership probabilities ≥ 0.80 were assigned to the corresponding subgroups and lines with membership < 0.80 were assigned to a mixed subgroup (Wang et al. 2008).
Results
Agro-morphological diversity
The ANOVA results showed that variability introduced by blocks was observed only for CD and RD, whereas for the other traits the block effects were not significant (Table 1). Data revealed high significant differences among checks for almost all the traits except cob (CD) and ear diameters (ED), ear length (EL), kernel row number (NKR) and ear number per plant (NEP) (Table 1). Moreover, the populations differed significantly for eight traits namely DS, DA, PH, RD, ERN, NEP and YP (Table 1).
The statistical parameters (mean, standard deviation, minimum, maximum and coefficient of variation) related to agro-morphological traits of populations are presented in Table 2. For earliness, in 63 and 68 days after sowing the earliest population (AGL) reached 50% anthesis and silking, respectively. On the contrary, the starting of anthesis and silking were delayed in IGR2 which was the latest population (96 and 98 days after sowing, respectively). The anthesis-silking interval (ASI) ranged from 1 to 4 days for more than 60% of the populations under study, and from 5 to 11 days for the remaining. Maximum plant and ear height (132.12 and 79.46 cm) were recorded in TLC and FEZ, whereas the minimum (45.79 and 14.47 cm) were observed in AGL and TWR, respectively (Table 2). Indeed, the highest population (TLC) recorded the highest yield (62.46 qt h−1), short ASI (2 days) and was late maturing (85 and 87 DA and DS, respectively). For ear related traits, high correlation was registered between ED and NKR. Indeed, population IGR2 showed largest ear (3.7 cm) as well as maximum number of ear rows (13.1). In addition, a correlation between EL and NKR was observed, with minimum values of 8.18 cm and 13.9 cm, respectively was registered in BNT population. The longest ear was observed in IZM (16.25 cm), while IGR4 showed the highest NKR. The ear number per plant (NEP) varied from 1.4 (AZA) to 3.10 (TMD2) with a mean of 2.25. The average 100-kernel weight (WHK) was 0.0231 kg ranging from 0.016 kg (IGR3) to 0.036 kg (MGR).
Analyzing the measured agro-morphological traits in maize germplasm, highly and positive correlations was found among most of the traits (Suppl. Table 3). The highest and positive correlation was registered between PH and EH (r = 0.95), followed by DA and DS (r = 0.94). Similarly, DA, DS, PH, EH, ED, EL, NKR and YP were highly and positively correlated with each other, highlighting a robust relationship among these traits. YP was weakly correlated with NEP (r = 0.28) (Suppl. Table 3).
The principal component analysis (PCA) based on 14 agro-morphological traits was performed to reduce the data set. Total variance explained by first two components was 55.44% (Suppl. Table 4). The first component (PC1) accounted for the highest rate 43.04% of total variation, in which the discriminatory traits were: DA, DS, PH and EH, EL and ED, NKR, ERN and YP (Suppl. Table 4). Cob diameter were positively correlated with second principal component (PC2) which explained 12.40% of total variation, whereas NEP and YP were negatively correlated (Suppl. Table 4).
The genotype-trait biplots (Fig. 1), based on the first two principal components, was also generated to evaluate the relationships among maize populations and all traits measured. In general, PC1 opposed late maturing and highest populations (TLC, IGR2, RGN, IZM, FEZ, GAG, EDD, and AGH) to the earliest and the shortest (TWR, AGL, AZA, SDY, BNT, TMD1 and MHF). In the same way, PC2 differentiate high yielding populations (MRG, TLC, DRR, GAG, IGR4 and AMR1) from low yielding (AGL, BFD, AZA, TNR, IGR3 and EGC) (Fig. 1).

Principal component analysis (PCA) of fifty-six maize populations based on agro-morphological traits
Genetic diversity
Polymorphism and allelic diversity
The polymorphisms among fifty-six maize populations were investigated by using 18 SSRs. These loci generated a total of 191 alleles with an average of 10.61 alleles per locus (Table 3). SSR marker bnlg1740 showed the highest number of alleles (24) followed by phi036 and umc-1335 (19 each), while the lowest number (3) was observed in umc-1265. The mean effective number of allele (Ne) and the shannon’s information index (I) were estimated to be 2.31 (ranging from 1.18 to 3.80 for phi 127 and umc-1335, respectively) and 0.90 (from 0.23 to 1.51 for phi127 and umc1335, respectively). Umc1335 exhibited the highest PIC (0.825), whereas the lowest was 0.14 (phi127). Across 18 SSR analyzed, nine showed PIC values greater than 0.6 (Table 3).
Among 191 alleles detected across 56 populations under study, nineteen unique alleles were detected in 13 SSR markers (Table 4). The highest number of private alleles (3) was detected using umc1222 marker, followed by phi036, umc1165, umc1225 and umc1424 with 2 alleles for each SSR. For the remaining, only one private allele each was found. These unique alleles were revealed in 14 populations out of 56, among them BAH showed the maximum number (3), while AMR1, AGH and TNR revealed 2 alleles for each population (Table 4).
The population’s parameters as allelic richness (Na) observed (Ho) and expected (He) heterozygosity as well as inbreeding coefficient (f) were also estimated and summarized in Table 4. The Na, which is defined as the average number of alleles detected across 18 loci, ranged from 5.16 (AMR1) to 3.11 (TLC). The highest expected and observed heterozygosity (He = 0.60 and Ho = 0.62) were registered for AMR1, whereas the lowest values (He = 0.37 and Ho = 0.38) were recorded for TLC. The average observed and expected heterozygosity for all accessions were 0.510 and 0.512 respectively. Overall inbreeding coefficient of the populations varied from − 0.017 (LHM) to 0.108 (FNG) (Table 4).
Population structure
The overall genetic differentiation (Fst = 0.22) indicated that 22% of total variation is due to differences among populations, while within population genetic variation accounted for 78%. Analysis of molecular variance (AMOVA) revealed highly significant differences among populations and within individuals (Table 5). Approximately, 79% of total variance occurred within individuals, while 20% was portioned among populations (Table 5). Structure analysis showed the Algerian populations separated in two genetic groups (K = 2) (Fig. 2). The percentage of individuals in each population belonging to each group were estimated (Table 4) and the cut-off probability for assignment to a cluster was assumed to be ≥ 0.8 according to the information provided by the Q-matrix (Pritchard et al. 2000). A set of 29 populations out of 56 showed values of proportion of membership greater than or equal to 0.8 and were therefore assigned to a specific group, while the remaining populations (27), with proportion below 0.8 were classified as intermediate (assigned to mixed group) (Table 4, Fig. 2). Group 1 (red) consisted of 10 populations which are mostly originated from Reggane and Zaouit Kounta located at east and central south of Adrar, respectively. Group 2 (green) gathered 19 populations, from In Salah located in north of Tamanrasset, Zaouit Kounta, Reggane and other localities; the remaining 27 populations made up the intermediate group (Fig. 2).

Bayesian individual clustering based on 18 SSR data of fifty-six maize populations as inferred by STRUCTURE (group 1 = red; group 2 = green). (Color figure online)
Cluster and principal coordinates analysis
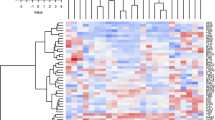
A dendrogram based on the genetic distance matrix of the SSR data was generated using the UPGMA algorithm (Fig. 3). The dendrogram revealed three large clusters in which the 56 maize populations were included. In cluster I, 44 populations were present and further divided into four sub-clusters (Ia, Ib, Ic and Id). Sub-cluster Ia included 12 accessions of which 5 were from mixed-group and 6 from group 2 as resulted by Bayesian clustering. Sub-cluster Ib contained also 12 populations, while sub-cluster Ic and Id gathered both 10 populations. Cluster II and III included 7 and 2 populations, respectively, which were mostly from group 1. The last 3 populations (ZRG, WTT and RGN) appeared as out-groups (Fig. 3). Finally, the Principal Coordinates Analysis (PCoA) was performed to cluster populations for their genetic similarity. Component 1 and 2 explained 12.16% and 10.10%, respectively (Fig. 4). The analysis was not able to distinguish populations for their geographical origin, as frequently observed (Fig. 4).

Unweight pair-group method of arithmetic averages (UPGMA) dendrogram generated from 18 SSR data showing relationships of fifty-six Algerian maize populations

Principal coordinate analysis (PCoA) based on 18 SSR data of fifty-six maize populations
Discussion
The present study investigated Algerian maize populations by a morphological and molecular characterization. Although morphological analysis for genetic diversity assessment presents many limitations as low polymorphism and influence of environment on phenotypic expression (Smith and Smith 1992), phenotypic traits were helpful in a preliminary evaluation of genetic diversity (Beyene et al. 2005a) and provided practical and critical information required to characterize genetic resources (Ignjatovic-Micic et al. 2015). On the other hand, molecular markers have been used successfully for genetic diversity and populations structure studies on maize (Dubreuil et al. 2006; Sharma et al. 2010; Cömertpay et al. 2012). Therefore, combination of both markers provided the best and effective conservation and management of genetic resources (Ristić et al. 2013).
The analysis of variance revealed a wide range of phenotypic traits variation. Maize populations from Sahara differed significantly for flowering time (DA, DS), ear traits (RD, ERN, and NEP), plant height, 100-kernel weight and grain yield. Despite the significant difference for flowering date, there were no significant differences for ASI. The highest population recorded the shortest ASI, highest crop yield and was late maturing, in agreement with previous study (Bolaños and Edmeades 1996), revealing that genotypes with short ASI exhibited higher yield, in agreement with Ngugi et al. (2013) who considered that short ASI is important trait in selection for increasing yield and Tabu et al. (2011) who demonstrated a correlation between ASI and yield loss. Relationships between different traits were investigated using the coefficient of correlation and PCA. The most significant correlations were found between days to anthesis and days to silking, plant and ear height, which is in agreement with similar findings in maize (Beyene et al. 2005b). Our results also revealed that many traits were significantly and positively correlated with each other. These results are in agreement with those obtained by Bolaños and Edmeades (1996); Mijangos-Cortés et al. (2007); Cömertpay et al. (2012) and Iqbal et al. (2015), which reported consistent correlations between agro-morphological traits in maize. Indeed, linked genes encoding for different phenotypic traits are the cause of this high correlation between different traits (Cömertpay et al. 2012; Iqbal et al. 2015). Thus, specific pairwise correlations between agronomic traits of interest should be taken into account by the breeders. However, the selection of flag traits should be performed carefully under different agro-climate conditions to avoid the influence of the environment in trait evaluation (Yücel et al. 2009).
The PCA was performed to classify the populations on the basis of the most discriminating traits. The results revealed two main components accounting for 55% of total variability. Among the traits analyzed, flowering time, plant and ear height, ear traits and grain yield played a crucial role in differentiating the populations. The PCA-based grouping of Algerian maize germplasm are in agreement with those obtained by Beyene et al. (2005b) and Hartings et al. (2008). Likewise, Mijangos-Cortés et al. (2007) and Jaric et al. (2010) reported that plant height and ear height, ear length and yield were also the most discriminating traits to identify maize populations in Eastern Serbia and Mexico, respectively. Similarly, Gouesnard et al. (1997) identified earliness and plant size as major traits contributing to the grouping of French maize accessions.
PCA are not able to classify the genotypes for their geographic origin. These results agree with those obtained by Hartings et al. (2008) and Sharma et al. (2010), who found that landraces from the same regions were not grouped together in the same cluster. The weak geographical relationship among populations can be explained by the extensive mixing of germplasm between fields due to pollen-mediated gene flow; while the exchange of seeds between farmers from distinct regions to meet their needs could be another reason (Beyene et al. 2005b; Cömertpay et al. 2012).
The phenotypic characterization revealed that Saharan maize germplasm display large amount of variability for quantitative traits. Therefore, grouping the populations for similar phenotypic traits could help the genetic improvement.
The molecular diversity of selected maize populations was also analyzed using fluorescent dye-labeled SSR markers with allele resolution using DNA-sequencer. The number of alleles and their frequencies at each locus were analyzed, as indicator of SSR polymorphism. In total, 191 alleles were detected among 56 populations, with an average of 10.62 allele per locus. The high number of alleles per locus found in this study is most likely attributable to the higher genetic diversity in the investigated germplasm. Accordingly, Adetimirin et al. (2008) revealed mean values of 9.7 alleles per SSR locus, after genotyping 17 inbred lines from west and central Africa. By contrast, Beyene et al. (2006b) and Choukan et al. (2006) reported an average of 4.9 alleles per locus analyzing 62 Ethiopian maize accessions and 58 Iranian inbred lines, respectively. The difference in the number of alleles between studies could be explained by several reasons; the methodologies used for detection of polymorphic markers; the size of the collection under study, the expected diversity or uniformity based on pedigrees and finally and mainly the SSR panel adopted (Heckenberger et al. 2002; Choukan et al. 2006; Adetimirin et al. 2008). Indeed, dinucleotide SSR markers generate higher number of alleles when compared to tri- and tetra- nucleotides repeats (Heckenberger et al. 2002).
A measure of genetic diversity of populations is the effective number of allele (Ne) which depend on the proportion of polymorphic loci, the allele number per locus as well as the uniform distribution of allele frequencies (Sharma et al. 2010). Across our populations, a mean of 2.31 was registered, which was lower to those recorded by Wasala and Prasanna (2012) and Qi-Lun et al. (2008) analyzing Indian maize accessions and Chinese landraces, respectively.
Among the SSR markers, umc1335 had the highest PIC (0.825) and a high number of alleles (19). Conversely, the low PIC value (0.14) with only 4 alleles was registered for phi127 which could be explained by the dependency of PIC to the number and frequency distribution of the alleles detected (Romero-Severson et al. 2001). The average PIC value (0.57) was higher than the values reported by Oppong et al. (2014) (0.50) and Nyaligwa et al. (2015) (0.51) using 20 and 30 SSR, respectively. Higher PIC values have been obtained in maize landraces from India (0.60) by using 42 SSRs (Sharma et al. 2010), Japan (0.69) using 60 SSRs (Enoki et al. 2002) and Turkish maize landraces (0.72) using 28 SSRs (Cömertpay et al. 2012).
The present study identified nineteen unique alleles, which are specific for single population, in 14 populations out of 56. The presence of such allele may be an indication of high rate of mutation in SSR loci (Henderson and Petes 1992). According to Wasala and Prasanna (2012), a selection of specific allele related to morphology and environment of adaptation can also occur. The average genetic diversity within accessions (He = 0.51) was higher than those reported by Noldin et al. (2016) (He = 0.48) and Aci et al. (2013) (He = 0.44) for Paraguayan and Algerians accessions, respectively; while Yao et al. (2007) reported the higher mean (He = 0.69) analyzing Chinese maize landraces. Total genetic diversity in our study (He = 0.61) was the same to that found in Mexican maize (0.61) by Reif et al. (2006) and lower than those registered by several other reports (Sharma et al. 2010; Eschholz et al. 2010).
The observed heterozygosity was lower than expected heterozygosity, which indicates an excess of homozygotes. Despite the outcrossing nature of maize, this phenomenon may be due to the maize cultivation in isolated Oases and a deficit of heterozygotes was already reported (Qi-Lun et al. 2008; Noldin et al. 2016). According to Snyder et al. (1985), who consider that a range from 0.15 to 0.25 of Fst values indicated high genetic divergence, our results revealed that maize populations have high level of genetic differentiation (Fst = 0.22) highlighting that the Saharan populations are being efficiently conserved by farmers. This result is confirmed by low rate of inbreeding revealing a high level of genetic identity of populations under study. Genetic differentiation (Fst = 0.22) among our populations was above those found for Chinese landraces (0.07) (Yao et al. 2007) and American Southern accessions (0.12) (Noldin et al. 2016). Higher Fst values were in contrast reported in Algerian (Aci et al. 2013) and Spanish (Romay et al. 2012) maize accessions, who reported Fst = 0.3. Fst mean was high (0.36 and 0.43, respectively) for Indian (Wasala and Prasanna 2012) and Swiss populations (Eschholz et al. 2008) and very high (0.93) among tropical sweet corn inbred lines (Kashiani et al. 2012). The dispersal of pollen and farmers exchanges are factors affecting the patterns of gene dispersion within and among populations (Loveless and Hamrick 1984). According to Da Silva et al. (2015) outcrossing plants usually maintain a consistent genetic variation within populations and low among them, in agreement with our result.
Analysis of molecular variance (AMOVA) is a suitable criterion to assess overall distribution of diversity within and among populations. In agreement with information provided by genetic differentiation coefficient, the AMOVA results showed a higher level of genetic variation within our populations than among them. Similar results were reported by Singode and Prasanna (2010) and Da Silva et al. (2015) analyzing 48 North Eastern Himalayan landraces using 41 SSRs and 31 popcorn accessions of the U.S germplasm at 30 SSR loci, respectively.
The analysis based on the STRUCTURE model identified two genetically differentiated groups, while the dendrogram depicted using the UPGMA algorithm grouped the accessions into 3 clusters. Despite the number of groups and clusters, UPGMA clustering was largely in agreement with the result of the Bayesian clustering since the cluster analysis based on both the molecular (UPGMA and STRUCTURE) and phenotypic data grouped the populations regardless their geographic origin. This populations structuring can be explained by a common genetic background despite the geographical and phenotypic divergence (N’Da et al. 2016). Indeed, the empirical selection for favorable alleles by farmers to meet their needs in term of adaptation to local conditions and the exchange of seeds among farmers from distinct regions could be the reason for this non-relatedness of maize populations from the same region (Cömertpay et al. 2012). The extensive mixing of germplasm between fields due to gene flow could be another reason (Thakur et al. 2016). Similarly, (Cömertpay et al. 2012), Noldin et al. (2016) and Thakur et al. (2016) did not found relationship between clusters based on molecular markers and geographic origin analyzing Turkish, Paraguayan and Himalayan maize landraces, respectively.
The results of the present study clearly revealed significant phenotypic and molecular diversity of the Saharan maize populations. Furthermore, this diversity among maize populations could be related to different plant response to abiotic stress since the geographic areas, where the landraces were collected, are frequently subjected to drought, severe high temperature and low soil fertility. Therefore, these promising maize populations could be potentially utilized for the introgression of adaptive traits, which may be found in extreme environments (Hawtin et al. 1996). The distribution of populations into morphologically and genetically similar groups should quicken the usefulness of these data to maize breeders.
References
Aci MM, Revilla P, Morsli A, Djemel A, Belalia N, Kadri Y, Khelifi-Saloui M, Ordás B, Khelifi L (2013) Genetic diversity in Algerian maize (Zea mays L.) landraces using SSR markers. Maydica 58:304–310
Aci MM, Lupini A, Mauceri A, Morsli A, Khelifi L, Sunseri F (2018) Genetic variation and structure of maize populations from Saoura and Gourara oasis in Algerian Sahara. BMC Genet 19:51
Adetimirin VO, Vroh-BI I, The C, Menkir A, Mitchell SE, Kresovich S (2008) Diversity analysis of elite maize inbred lines adapted to west and central Africa using SSR markers. Maydica 53:143–149
Adeymo O, Menkir A, Melaku G, Omidiji O (2011) Genetic diversity assessment and relationship among tropical-yellow endosperm maize inbred line using SSR markers. Maydica 56:1703–1709
Betrán FJ, Beck D, Banziger M, Edmeades GO (2003) Genetic analysis of inbred and hybrid grain yield under stress and non-stress environments in tropical maize. Crop Sci 43:807–817. https://doi.org/10.2135/cropsci2003.8070
Beyene Y, Botha AM, Myburg AA (2005a) A comparative study of molecular and morphological methods of describing genetic relationships in traditional Ethiopian highland maize. Afr J Biotechnol 4:586–595
Beyene Y, Botha AM, Myburg AA (2005b) Phenotypic diversity for morphological and agronomic traits in traditional Ethiopian highland maize accessions. S Afr J Plant Soil 22:100–105. https://doi.org/10.1080/02571862.2005.10634689
Beyene Y, Botha AM, Myburg A (2006a) Genetic diversity among traditional Ethiopian highland maize accessions assessed by simple sequences repeat (SSR) markers. Gen Res Crop Evol 53:1579–1588. https://doi.org/10.1007/s10722-005-8509-y
Beyene Y, Botha AM, Myburg AA (2006b) Genetic diversity in traditional Ethiopian highland maize accessions assessed by AFLP markers and morphological traits. Biodiv Conserv 15:2655–2671. https://doi.org/10.1007/s10531-005-5407-8
Bolaños J, Edmeades GO (1996) The importance of the anthesis-silking interval in breeding for drought tolerance in tropical maize. Field Crop Res 48:65–80. https://doi.org/10.1016/0378-4290(96)00036-6
Carimi F, Mercati F, De Michele R, Fiore MC, Riccardi P, Sunseri F (2011) Intra-varietal genetic diversity of the grapevine cultivar Nero d’Avola as revealed by microsatellite markers. Genet Resour Crop Evol 58:967–975. https://doi.org/10.1007/s10722-011-9731-4
Chen JP, Xu WW, Velten J, Xin ZG, Stout J (2012) Characterization of maize inbred lines for drought and heat tolerance. J Soil Water Conserv 67:354–364. https://doi.org/10.2489/jswc.67.5.354
Choukan R, Hossinzade A, Ghanadha MR, Warburton ML, Talei A, Mohammadi SA (2006) Use of SSR data to determine relationships and potential heterotic groupings within medium to late maturing Iranian maize inbred lines. Field Crop Res 95:212–222. https://doi.org/10.1016/j.fcr.2005.02.011
CIMMYT and IBPGR (1991) Descriptors of maize. CIMMYT/IBPGR, Rome, p 88
Cömertpay G, Baloch FS, Kilian B, Ülger AC, Özkan H (2012) Diversity assessment of Turkish maize landraces based on fluorescent labelled SSR markers. Plant Mol Biol Rep 30:261. https://doi.org/10.1007/s11105-011-0332-3
Djemel A, Revilla P, Hanifi-Mekliche L, Malvar RA, Alvarez A, Khelifi L (2012) Maize (Zea mays L.) from the Saharan oasis: adaptation to temperate areas and agronomic performance. Genet Resour Crop Evol 59:1493–1504. https://doi.org/10.1007/s10722-011-9778-2
Dubreuil P, Warburton ML, Chastanet M, Hoisington D, Charcosset A (2006) More on the introduction of temperate maize into Europe: large-scale bulk SSR genotyping and new historical elements. Maydica 51:281–291
Enoki H, Sato H, Koinuma K (2002) SSR analysis of genetic diversity among maize inbred lines adapted to cold regions of Japan. Theor Appl Genet 104:1270–1277
Eschholz TW, Peter R, Stamp P, Hund A (2008) Genetic diversity of Swiss maize assessed with individuals and bulks on agarose gels. Genet Resour Crop Evol 55:971–983
Eschholz TW, Stamp P, Peter R, Leipner J, Hund A (2010) Genetic structure and history of Swiss maize (Zea mays L. ssp. mays) landraces. Genet Resour Crop Evol 57:71–84. https://doi.org/10.1007/s10722-009-9452-0
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier LL, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Federer WT (1956) Augmented (or hoonuiaku) designs. Hawaii Plant Rec 55:128–191
Franco J, Crossa J, Ribaut JM, Betran J, Warburton ML, Khairallah M (2001) A method for combining molecular markers and phenotypic attributes for classifying plant genotypes. Theor Appl Genet 103:944–952. https://doi.org/10.1007/s001220100641
Gouesnard B, Dallard J, Panouillé A, Boyat A (1997) Classification of French maize populations based on morphological traits. Agronomie 17:491–498
Govindaraj M, Vetriventhan M, Srinivasan M (2015) Importance of genetic diversity assessment in crop plants and its recent advances: an overview of its analytical perspectives. Genet Res Int 2015:431487
Hartings H, Berardo N, Mazzinelli GF, Valoti P, Verderio A, Motto M (2008) Assessment of genetic diversity and relationship among maize (Zea mays L.) Italian landraces by morphological traits and AFLP profiling. Theor Appl Genet 117:831–842. https://doi.org/10.1007/s00122-008-0823-2
Hawtin G, Iwanaga M, Hodgkin T (1996) Genetic resources in breeding for adaptation. Euphytica 92:255–266. https://doi.org/10.1007/BF00022853
Heckenberger A, Melchinger AE, Ziegle JS, Joe LK, Häuser JD, Hutton M, Bohn M (2002) Variation of DNA among accessions within maize inbred lines and implications for identification of essentially derived varieties. I. Genetic and technical sources of variation in SSR data. Mol Breed 10:181–191. https://doi.org/10.1023/A:1026040007166
Henderson ST, Petes TD (1992) Instability of simple sequence DNA in Saccharomyces cerevisiae. Mol Cell Biol 12:2749–2757
Hoxha S, Shariflou MP, Sharp P (2004) Evaluation of genetic diversity in Albanian maize using SSR markers. Maydica 49:97–103
Ignjatovic-Micic D, Ristic D, Babic V, Andjelkovic V, Vancetovic J (2015) A simple SSR analysis for genetic diversity estimation of maize landraces. Genetika 47:53–62
Iqbal J, Shinwari ZK, Rabbani MA (2015) Maize (Zea mays L.) germplasm agro-morphological characterization based on descriptive, cluster and principal component analysis. Pak J Bot 47:255–264
Jaric JK, Prodanovic S, Iwarsson M, Minina A (2010) Diversity of maize (Zea mays L.) landraces in Eastern Serbia: morphological and storage protein characterization. Maydica 55:231–238
Kashiani P, Saleh G, Panandam JM, Abdullah NAP, Selamat A (2012) Molecular characterization of tropical sweet corn inbred lines using microsatellite markers. Maydica 57:154–163
Kumar S, Stecher G, Tamura K (2015) MEGA/: molecular evolutionary genetics analysis version 7.0 for bigger dataset. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Legesse BW, Myburg AA, Pixley KV, Botha AM (2007) Genetic diversity of African maize inbred lines revealed by SSR markers. Hereditas 144:10–17. https://doi.org/10.1111/j.2006.0018-0661.01921.x
Lewontin RC (1972) Testing the theory of natural selection. Nature 236:181–182
Loveless MD, Hamrick JL (1984) Ecological determinates of genetic structure in plant populations. Ann Rev Ecol System 15:65–95. https://doi.org/10.1146/annurev.es.15.110184.000433
McCann JC (2005) Maize and grace Africa’s encounter with a new world crop. Harvard University Press, Cambridge, pp 1500–2000
Mercati F, Leone M, Lupini A, Sorgonà A, Bacchi M, Abenavoli MR, Sunseri F (2013) Genetic diversity and population structure of a common bean (Phaseolus vulgaris L.) collection from Calabria (Italy). Genet Res Crop Evol 60:839–852. https://doi.org/10.1007/s10722-012-9879-6
Messmer MM, Melchinger AE, Hermann RG, Boppenmaier J (1993) Relationship among early European maize inbreds. II. Comparison of pedigree and RFLP data. Crop Sci 33:944–950. https://doi.org/10.2135/cropsci1993.0011183X003300050014x
Mijangos-Cortés JO, Corona-Torres T, Espinosa- Victoria D, Muñoz-Orozco A, Romero-Peñaloza J, Santacruz-Varela A (2007) Differentiation among maize (Zea mays L.) landraces from the Tarasca Mountain Chain, Michoacan, Mexico and the Chalqueno complex. Genet Resour Crop Evol 54:309–325. https://doi.org/10.1007/s10722-005-4775-y
N’Da HA, Akanvou L, Pokou ND, Akanza KP, Kouakou CK, Zoro BIIA (2016) Genetic diversity and population structure of maize landraces from Côte d’Ivoire. Afr J Biotech 15:2507–2516
Nei M, Li W (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76:5269–5273
Ngugi K, Cheserek J, Muchira C, Chemining’wa G (2013) Anthesis to silking interval usefulness in developing drought tolerant maize. J Renew Agric 1:84–90
Noldin O, Revilla P, Ordás B (2016) Genetic diversity of the floury race of maize Avati Morotî from the Guaraní tribe in Paraguay. Span J Agric Res 14:e0707
Nyaligwa L, Hussein S, Amelework B, Ghebrehiwot H (2015) Genetic diversity analysis of elite maize inbred lines of diverse sources using SSR markers. Maydica 60:1–8
Oppong A, Bedoya CA, Ewool MB, Asante MD, Thompson RN, Adu-Dapaah H, Lamptey JNL, Ofori K, Offei SK, Warburton ML (2014) Bulk genetic characterization of Ghanaian maize landraces using microsatellite markers. Maydica 59:1–8
Peakall R, Smouse PE (2006) GenAlEx6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Prasanna BM (2012) Diversity in global maize germplasm: characterization and utilization. J Biosci 37:843–855
Pritchard JK, Stephens M, Donelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Qi-Lun Y, Ping F, Ke-Cheng K, Guang-Tang P (2008) Genetic diversity based on SSR markers in maize (Zea mays L.) landraces from Wuling mountain region in China. J Genet 87:287–291
R Development Core Team (2008) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org
Rebourg C, Gousnard B, Charcosset A (2001) Large scale molecular analysis of traditional European maize populations: relationships with morphological variation. Heredity 86:574–587
Reif JC, Warburton ML, Xia XC, Hoisington DA, Crossa J, Taba S, Muminovic J, Bohn M, Frisch M, Melchinger AE (2006) Grouping of accessions of Mexico races of maize revisited with SSR markers. Theor Appl Genet 113:177–185. https://doi.org/10.1007/s00122-006-0283-5
Ristić D, Babić V, Andelković V, Vancetović J, Miladenović Drinić S, Ignjatović-Micić D (2013) Genetic diversity in maize dent landraces assessed by morphological and molecular markers. Genetika 45:811–824
Romay MC, Butrón A, Ordás A, Revilla P, Ordás B (2012) Effects of recurrent selection on the genetic structure of two broad base Spanish maize synthetics. Crop Sci 52:1493–1502. https://doi.org/10.2135/cropsci2011.10.0552
Romero-Severson J, Smith JSC, Ziegle J, Hauser J, Joe L, Hookstra G (2001) Pedigree analysis and haplotype sharing within diverse groups of Zea mays L. inbreds. Theor Appl Genet 103:567–574. https://doi.org/10.1007/PL00002911
Schuelke M (2000) An economic method for the fluorescent labelling of PCR fragments. Nat Biotechnol 18:233–234. https://doi.org/10.1038/72708
Sharma L, Prasanna BM, Ramesh B (2010) Analysis of phenotypic and microsatellite-based diversity and population genetic structure of maize landraces in India, especially from the North-East Himalayan region. Genetica 138:619–631
Shiferaw B, Prasanna BM, Hellin J, Bänziger M (2011) Crops that feed the world 6. Past successes and future challenges to the role played by maize in global food security. Food Secur 3:307–327
Silva TA, Cantagalli LB, Saavedra J, Lopes AD, Mangolin CA, Pires MF, Machado S, Scapim CA (2015) Population structure and genetic diversity of Brazilian popcorn germplasm inferred by microsatellite markers. Electron J Biotech 18:181–187
Singode A, Prasanna BM (2010) Analysis of genetic diversity in the North-Eastern Himalayan (NEH) maize landraces of India using microsatellite markers. J Plant Biochem Biotech 19:33–41
Smith JSC, Smith OS (1992) Finger printing crop varieties. Adv Agron 47:85–140
Sneath PHA, Sokal RR (1973) Numerical taxonomy: the principles and practice of numerical classification. Freeman and Company, San Francisco
Snyder LA, Freifelder D, Hartl DL (1985) General genetics. Jones and Bartlett, Boston
Tabu I, Munyiri S, Pathak R (2011) Phenotypic characterization of local maize landraces for drought tolerance in Kenya. In: Bationo A, Waswa B, Okeyo J, Maina F, Kihara J (eds) Innovations as key to the green revolution in Africa. Springer, Dordrecht
Thakur N, Prakash J, Thakur K, Sharma JK, Kumari R, Rana M, Lata S (2016) Genetic diversity and Structure of maize accessions of North Western Himalayas based on morphological and molecular markers. Proc Natl A Sci India B 87:1385–1398
Wang RH, Yu YT, Zhao JR, Shi YS, Song YC, Wang TY, Li Y (2008) Population structure and linkage disequilibrium of a mini core set of maize inbred lines in China. Theor Appl Genet 117:1141–1153. https://doi.org/10.1007/s00122-008-0852-x
Warburton ML, Xia X, Crossa J, Franco J, Melchinger AE, Frisch M, Bohn M, Hoisington D (2002) Genetic characterization of CIMMYT maize inbred lines and open pollinated populations using large scale fingerprinting methods. Crop Sci 42:1832–1840. https://doi.org/10.2135/cropsci2002.1832
Wasala SK, Prasanna BM (2012) Microsatellite markers-based diversity and population genetic analysis of selected lowland and mid-altitude maize landraces accessions of India. J Plant Biochem Biotech 22:392–400. https://doi.org/10.1007/s13562-012-0167-5
Wietholter P, de Melo Cruz, Sereno MJ, de Freitas Terra T, dos Anjos Delmar, Silva S, Barbosa Neto JF (2008) Genetic variability in corn landraces from Southern Brazil. Maydica 53:151–159
Yao Q, Fang P, Kang K, Pan G (2007) Genetic diversity based on SSR markers in maize (Zea mays L) landraces from Wuling mountain region in China. J Genet 87:287–291
Yücel C, Hizli H, Firincioglu HK, Cil A, Anlarsal AE (2009) Forage yield stability of common vetch (Vicia sativa L.) genotypes in the Cukurova and GAP Regions of Turkey. Turk J Agric For 33:119–125
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Belalia, N., Lupini, A., Djemel, A. et al. Analysis of genetic diversity and population structure in Saharan maize (Zea mays L.) populations using phenotypic traits and SSR markers. Genet Resour Crop Evol 66, 243–257 (2019). https://doi.org/10.1007/s10722-018-0709-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-018-0709-3