
Abstract
The Mexican fruit fly, Anastrepha ludens, is a highly significant agricultural pest species that has been genetically transformed with a piggyBac-based transposon vector system using independent vector and transposase helper plasmids. Minimum estimated germ-line transformation frequencies were approximately 13–21% per fertile G0 individual, similar to previously reported frequencies using single vector-helper plasmids. Two vector constructs were tested with potential importance to transgenic strain development for mexfly biological control. The first allows post-integration stabilization of a transposon-vector by deletion of a terminal sequence necessary for mobilization. The complete pB[L1-EGFP-L2-DsRed-R1] vector was integrated into the Chiapas wild type strain with subsequent deletion of the L2-DsRed-R1 sub-vector carrying the piggyBac 3′ terminal sequence. Quality control tests for three of the stabilization vector lines (previous to stabilization) assessed viability at all life stages, fertility, adult flight ability, and adult male sexual competitiveness. All three transgenic lines were less fit compared to the wild strain by approximately 5–10% in most tests, however, there was no significant difference in sexual competitiveness which is the major prerequisite for optimal strain release. The second vector, pB[XL-EGFP, Asß2-tub-DsRed.T3], has the DsRed.T3 fluorescent protein reporter gene regulated by the A. suspensa Asß2-tubulin promoter, that resulted in testis and sperm-specific DsRed fluorescence in transgenic male mexflies. Fluorescent sperm bundles were unambiguously observed in the spermathecae of non-transgenic females mated to transgenic males. One transgenic line apparently had a male-specific Y-chromosome insertion, having potential use for sexing by fluorescent-embryo sorting. All transgenic lines expressed easily detectable and stable fluorescence in adults allowing their identification after trapping in the field.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Mexican fruit fly, Anastrepha ludens, is one of the most economically important pest species attacking fruits such as oranges and mangoes in Mexico, that has made inroads within the continental U.S. in south Texas. The mexfly has been subject to regulation and control programs in Mexico since 1992, conducted by The National Campaign of Fruit Flies established by the federal government, which has established the concept of fly free zones for approximately 47% of the country. This has allowed the export of fruits with an estimated value of 34.4 million dollars (Gutiérrez-Ruelas and Santiago-Martínez 2008), which has been achieved by large-scale suppression, eradication and preventive control programs based on the Sterile Insect Technique (SIT) (Knipling 1955). Mexfly SIT relies on the large-scale release of radiation-sterilized insects provided by the Moscafrut mass rearing facility producing 190 million A. ludens flies per week (Gutiérrez-Ruelas and Santiago-Martínez 2008). SIT for mexfly and other tephritid pests, as well as other pests of economic and medical importance, is hindered by inefficiencies related to cost and sub-optimal effectiveness. Specifically, is the need to sterilize and release females as well as males in the absence of a sexing system in early development, use of harmful and inefficient fluorescent powders for marking, and radiation-induced sterility that diminishes fitness and viability. Classical genetic manipulations, including use of mutations and chromosomal rearrangements, have provided some strains in other tephritid species that allow sexing for male only development, and marking for field detection (Robinson et al. 1999). However, development of these strains has been labor-intensive and not easily transferable to other species. The use of recombinant DNA to create transgenic strains has allowed the development of new strains for genetic marking, sexing and conditional lethality/sterility in some tephritid species, the medfly in particular (Handler 2002, 2004; Alphey 2002; Robinson et al. 2004). The mexfly has been similarly transformed with piggyBac vectors (Condon et al. 2007b) and we describe efforts for continued development of this methodology using piggyBac transgene vectors for improved transformation of the species, and new vectors that allow post-integration stabilization of the transgenes and sperm-marking.
Materials and methods
Wild type insects
The wild type Chiapas strain, was collected from fruits proximal to the Moscafrut facility in Metapa de Dominguez, Chiapas, México, and has been maintained there for more than 20 generations (Zepeda-Cisneros et al. 2009). The wild A. ludens females used in sexual competitive tests were obtained from infested Citrus aurantium L. collected in the Soconusco Region, Chiapas, México, and males were obtained from Moscafrut, which has produced artificially reared flies since 1992 (Rull Gabayet et al. 1996).
Plasmids
The piggyBac vector, pB[L1-EGFP-L2-DsRed-R1], is a stabilization vector (see Handler et al. 2004) having an internal piggyBac 5′ (left arm; L2) terminal sequence inserted in between the PUb-nls-EGFP and PUb-DsRed.T3 marker sequences, and in direct tandem orientation with the external 5′ terminal (L1) sequence (see Fig. 2). The vector was constructed by digesting pB[PUb-DsRed1] (Handler and Harrell 2001a) just upstream (or 5′) to its piggyBac 5′ terminal sequence and inserting the PUb-nls-EGFP marker and a linked piggyBac 5′ terminal sequence from pB[PUb-nls-EGFP] (Handler and Harrell 2001b), creating a L1-PUb-nls-EGFP-L2-PUb-DsRed1-R1 configuration (the piggyBac 3′ terminal sequence, or right arm designated as R1). This vector can integrate using the L1 and R1 termini with resultant transformants marked with both EGFP and DsRed, or just the L2 and R1 termini, having only the DsRed marker.
The pB[XL-EGFP, Asß2tub-DsRed.T3] vector has been described (Zimowska et al. 2009), but briefly, the A. suspensa ß2-tubulin promoter region (1.3 kb upstream to the translation initiation site) was amplified with XhoI/BamHI sites and inserted into the p[DsRed.T3-N1] multiple cloning site 5′ to DsRed.T3 (Bevis and Glick 2002). The Asß2tub/DsRed.T3 fragment was ligated into the BglII and EcoRV sites within the piggyBac vector, pB[XLII-PUbEGFP] to create pB[XL-EGFP/Asß2tub-DsRed.T3]. The helper plasmid, phsp-pBac, having the piggyBac transposase gene under hsp70 regulation, with a deletion of the 5′ terminal sequence, was described previously (Handler and Harrell 1999).
Germ-line transformation
Embryo injections used procedures developed for the Mediterranean fruit fly (Handler et al. 1998), that were modified from standard Drosophila procedures. However, due to the longer developmental time for mexfly compared to other transformed tephritids, eggs were collected within 30–40 min of oviposition and then allowed to continue developing for 50–60 min at room temperature before dechorionation. Eggs were dechorionated in 1.6% hypochlorite solution followed by several washes in 0.02% Triton-X 100 in water, placed on double-stick tape, and desiccated in room-air and then overlayed with Halocarbon 700 oil in which they were injected. DNA mixtures had vector:helper concentrations of 500:300 μg/ml in injection buffer (5 mM KCl; 0.1 sodium phosphate pH 6.8). Injected G0 eggs were placed in an oxygenated and humidified tissue culture chamber at 23–25°C and heat shocked at 37°C for 1 h at 16–20 h after injection (heat-shock treatment is now considered optional). Enclosed G0 adults were backcrossed in small groups to Chiapas wild type host flies, with resulting G1 adult progeny examined under epifluorescence optics for either EGFP or DsRed.
PCR analysis
Genomic insertion site sequences at the 5′ and 3′ of ends of the integrated pB[L1-EGFP-L2-DsRed-R1] vector were determined by standard inverse PCR protocols. Genomic DNA, isolated from parental lines and stabilized lines using DNAzol (Molecular Research Center), was digested with HaeIII and ligated overnight at 16°C. PCR amplifications were performed on ligated DNA with primers 122R and 139F (see Table 3 for primer sequences) for the 5′ end and 144F and 151R for the 3′ end with Expand Long Template DNA polymerase (Roche Applied Science) using the following cycling conditions: initial denaturation at 95°C for 5 min, followed by 35 cycles at 95°C for 30 s, 55°C for 30 s and 68°C for 2 min with a final extension at 68°C for 7 min. Products were cloned into the TOPO TA vector pCR 2.1 (Invitrogen) and sequenced with vector primers using BigDye terminator chemistry (Applied Biosystems) to determine the genomic sequences at the insertion site. To further verify the insertion sites, PCR amplifications were performed with primers for the proximal genomic insertion site region and within the inserted vector sequence. For line 11M3, the 5′ end was amplified using primers 534F and 122R and the 3′ end using primers 515R and 144F. PCR products were analyzed on a 1% agarose gel, and for some, excised and extracted for subcloning and sequencing.
Quality control tests
Biological attributes
Fresh eggs were collected from cages containing 100 male and female pairs of each transgenic line or wild type control insects from the Chiapas strain. One-hundred eggs per replicate were placed on wet blotting paper in a humidified petri dish and incubated at 26°C. The maximum hatch was recorded after 6 days of egg collections as an estimate of fertility. Survival to various life stages was tested by placing 100 newly hatched first instar larvae on larval medium and recording the number of surviving larvae, pupae and adults, with these percentages used to calculate overall fitness (McCombs et al. 1993).
To estimate the quality of adult fitness flight ability tests were performed. The procedures for this test were made using the standard protocol described in the Product Quality Control and Shipping Procedures for Sterile Mass-Reared Tephritid Fruit Flies (see FAO/IAEA/USDA 2003). Briefly, 2 days before adult emergence 100 pupae were placed inside a black powdered tube allowing only flying adults to escape. After emergence, the contents of the tube were counted which included unemerged adults (pharate pupae), partially emerged adults, emerged but deformed adults, and adults unable to fly. ‘Fliers’ is the percentage of adult fliers/100 pupae (all pupae tested), and ‘Flight Index’ is the percentage fliers/emerged adults (all pupae minus unemerged and partially emerged adults). For each transgenic and control strain 15 replicates of this experiment were performed.
Sexual competitiveness
This test was performed under laboratory conditions within a small cage (30 × 30 × 30 cm) containing a mango tree branch. Ten transformed and 10 wild mass-reared males were placed in the cage after which 10 wild females from the field were introduced. The type of mating pair and mating number were observed and the proportion of transformed males mating relative to the total number of wild female matings was calculated as the relative sterility index (RSI; Orozco-Dávila et al. 2007). Five replicates were made for each line tested.
Data analysis
Proportions were arcsine √x transformed for the analyses (Zar 1999). A repeated measures multivariate ANOVA was used to analyze the biological attributes, flight ability and sexual competitive tests using the software StatView from SAS, version 5.0.
The Relative Sterility Index (RSI) measures the proportion of wild females mating with laboratory males. Under conditions where an equal number of transformed and mass reared males are released, the RSI indicates higher (>0.50) or lower (<0.50) competitiveness of sterile (or transformed) males compared with wild males in mating with wild females (McInnis et al. 1996).
Results
pB[L1-EGFP-L2-DsRed-R1] stabilization vector transformants
The first germ-line transformation was performed using the piggyBac vector, pB[L1-EGFP-L2-DsRed-R1], designed to allow post-integration removal of the DsRed marker and the single 3′ piggyBac terminus (R1) (see Fig. 2 for schematic representation of the vector). Since piggyBac transposition depends on intact 5′ and 3′ terminal sequences, loss of the 3′ terminus is expected to result in genomic stabilization of the remaining vector sequences as demonstrated previously (Handler et al. 2004). The vector was injected into 1,834 eggs from the Chiapas wild type strain, from which 355 larvae hatched with 185 surviving to the pupal stage (Table 1). Of these pupae, 138 G0 adults emerged, including 65 males and 73 females, that were backcrossed to wild type individuals in 23 small groups (3 G0 males mated to 5 wild type females and 5 G0 females mated to 3 wild type males) (Supplementary Table 1). From 13 G0 male groups, 10 yielded red/green fluorescent G1 progeny (Fig. 1a) with 7 of these groups also yielding, in addition, only red fluorescent progeny, and one group having no progeny. From 10 G0 female groups, five yielded red/green fluorescent G1 progeny with four of these groups also yielding only red fluorescent progeny, and two groups having no progeny. Thus, from the 20 fertile group matings, 15 yielded G1 transgenic flies totalling 392 G1 adults out of 3,854 screened.

Brightfield and epifluorescent images of a an adult A. ludens individual transformed with pB[L1-EGFP-L2-DsRed-R1] under brightfield (left), EGFP epifluorescence (center), and DsRed epifluorescence (right); and b a transgenic individual stabilized by remobilization of the L2-DsRed-R1 transgene sequences under EGFP epifluorescence (left), and DsRed epifluorescence (right), showing the loss of this marker. Co-expression of EGFP and DsRed appear orange in color using the FITC/EGFP filter
Since G0 flies were not tested for transformation events individually, the transformation frequency can only be estimated, as it was for the first mexfly transformations. Using the most conservative assumptions, it was assumed that all G0 flies were fertile, and that only single integration events occurred in independent G0 flies in groups yielding G1 transformants. For groups yielding red/green and red-only G1 transformants, single independent integration events must have occurred for each phenotype in different G0 flies, yielding at least two events per group. Thus, a minimum number of independent integrations would be 15 for red/green transformants and 11 for red-only transformants. This yields a total of 26 independent transformant lines from 125 G0 individuals (deducting 13 for groups yielding no progeny), resulting in a minimum transformation frequency of approximately 21% per fertile G0. This would be the minimal transformation frequency possible, with the actual frequency likely being higher due to G0 infertility, which is common (often observed at 50%; see Handler and Harrell 2001b).
A total of 26 red-green lines, having the complete vector integration were established, and after outcrosses all but one was determined have an autosomal insertion, with one line having an X-linked integration. Only a few lines expressed fluorescence in early stages, possibly the result of enhanced gene expression due to genomic position effects.
Post-integration stabilization
To stabilize the vector post-integration, three red-green lines, 1M1, 11M3, and 3F6, were selected for re-mobilization of the L2-DsRed-R1 sub-vector sequence. Flies homozygous for the transgene were outcrossed to the Chiapas wild strain, with resulting heterozygous F1 embryos injected with 500 ng/μl of phsp-pBac helper, reared to adulthood and outcrossed to Chiapas in small mating groups. F2 progeny were then examined for fluorescent phenotypes as adults (Table 2). All three lines produced progeny having non-fluorescent wild type phenotypes (not shown), as well as the red/green, green-only (Fig. 1b) and red-only (not shown) fluorescent phenotypes. The green-only fluorescent individuals were presumed to have stabilized L1-EGFP transgene sequences due to the remobilization of the L2-DsRed-R1 sequences. The origin of the red-only fluorescent individuals is more ambiguous, though it is likely these resulted from transposition of the L2-DsRed-R1 sub-vector to another chromosome that segregated solely with chromosomes having either no transgene insertion, or another L2-DsRed-R1 sequence. In some cases, it is possible that red-green fluorescent individuals had a stabilized transgene that segregated with a remobilized DsRed transgene, but this was not tested. Putative stabilized green-only sub-lines were selected for further evaluation.
PCR analysis
The 5′ and 3′ adjacent insertion site genomic sequences for pB[L1-EGFP-L2-DsRed-R1] in lines 11M3 and 3F6 were determined by inverse PCR, indicating duplicated TTAA insertion resulting from piggyBac-mediated insertions (Fig. 2; data not shown for 3F6). The original 1M1 strain was lost previous to molecular analysis and not sequenced. Primers to proximal genomic sequences were then used with internal vector primers to assess vector integrity for the initial vector integration, and then to reaffirm proper remobilization (i.e. deletion) of the L2-DsRed-R1 sequence (Table 3; Fig. 2 and Supplementary Fig. 1). The data for line 11M3 is shown including PCR product sizes consistent with predicted sizes, with diagnostic fragments sequenced to confirm loss of the L2-DsRed-R1 sub-vector (primers 387F/515R) with the L1-EGFP transgene sequence remaining intact within the genome (primers 534F/122R).

PCR analysis of the integrated pB[L1-EGFP-L2-DsRed-R1] vector in line 11M3, and the stabilized L1-PUbEGFP transgene after remobilization of the L2-DsRed-R1 sub-vector (see “Materials and methods” for PCR protocols and primer sequences). The relative forward (F) and reverse (R) primer position sites used for inverse and direct PCR (see Table 3 for primer sequences) are indicated above the diagram of the pB[L1-EGFP-L2-DsRed-R1] integrated vector (not to scale) with the excised L2-DsRed-R1 sequence indicated by the bracket above. Relative positions of HaeIII sites used to generate inverse PCR products are shown. Below the vector diagram is an ethidium bromide stained agarose gel with PCR products generated to assess the integrity of the vector insertion, and to verify that the re-mobilization event, consistent with loss of DsRed, had occurred in the expected manner. See Table 3 for predicted and observed PCR product sizes and Supplementary Fig. 1 for sequences of PCR products extracted from lanes 1, 3, 5, and 9
Fluorescent phenotype analysis
Individuals from the stabilized green-only fluorescent sub-lines were inbred to homozygosity and examined for fluorescent intensity (Fig. 1b). Unambiguous fluorescence was detected in all individuals from the thoracic flight muscles, with slightly more fluorescence extending into the posterior abdomen in line 3F6, followed in intensity in lines 11M3 and 1M1. Persistence of fluorescence after death was tested in 1 d adult decapitated flies with subsequent maintenance of the bodies in a petri dish under dry conditions. Fluorescence in all lines was detected for at least 2 months after death under these conditions (data not shown).
Quality control tests
For biological attribute tests viability at all life stages was assayed in the three pB[L1-EGFP-L2-DsRed-R1] transgenic lines compared to the wild Chiapas host strain (Table 4). Fertility was comparable in all transgenic lines ranging from approximately 85% in 11M3 to nearly 89% in 3F6 compared to 96% in the Chiapas strain, with larval viability ranging from 82.5 to 86.5% in the transgenics compared to 91% in Chiapas. Pupal and adult survival rates were more equivalent in the transgenic and wild Chiapas strains, though overall fitness was significantly lower in the transgenic lines. Notably line 1M1, which could not be maintained in lab rearing, showed the highest relative overall fitness of the transgenic lines.
The flight ability test (Table 5) indicated that line 11M3 had the lowest level of adult emergence close to 90%, compared to nearly 95% in both 1M1 and 3F6, and 98.5% in Chiapas. More partially emerged and deformed flies occurred in the transgenics, and the percent fliers ranged from approximately 82 to 88% in transgenics compared to 97.5% in Chiapas. The flight index ranged from 0.87 to 0.93 in transgenics compared to 0.99 in Chiapas.
Sexual competitive tests indicated, on average, that 11M3 and Chiapas had a nearly identical RSI, while the RSIs for 1M1 was slightly higher and slightly lower for 3F6 compared to Chiapas (Fig. 3). However, these differences were not statistically significant, and thus no significant differential tendency for females to choose males from a specific strain was observed.

Comparisons of the relative mating ability of transgenic and wild type males based on the relative sterility index (RSI). See “Materials and methods” for details
pB[XL-EGFP/Asß2t-DsRed.T3] vector for sperm marking
The second vector tested, pB[XL-EGFP/Asß2t-DsRed.T3], was previously transformed into A. suspensa (Zimowska et al. 2009), and exhibits polyubiquitin-regulated EGFP expression in all tissues, though most highly visible from the thoracic flight muscles, in addition to sperm-specific DsRed fluorescence, driven by a spermatocyte-specific ß2-tubulin promoter from A. suspensa. The vector was injected into the 313 embryos from which 65 hatched and 49 larvae survived to pupation (Table 1). Of these pupae, 38 adults emerged including 18 males and 20 females. The G0 progeny were crossed to wild type flies in 11 small groups from which five groups yielded transgenic progeny (Supplementary Table 2). Assuming a 100% fertility rate and only one transformation event per group mating, a minimum transformation frequency of approximately 26% per fertile G0 was estimated.
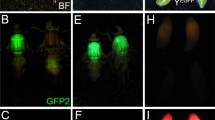
Of the G1 fluorescent progeny, 5 males were selected with strong red fluorescence expression in the testes (Fig. 4a), which were backcrossed to wild type females, and established as homozygous lines in successive generations. In addition, one line was presumed to be Y-linked due to EGFP expression completely limited to males (Fig. 5), however, testis-specific DsRed expression was not observed in this line presumably due to position effect suppression. This transgene was subsequently re-mobilized to an autosome where testis-specific DsRed expression is now observed (data not shown). As with the A. suspensa transformants, DsRed fluorescence could be observed through the posterior abdominal cuticle in males, that was shown to be specific to testes after their dissection, and sperm bundles after their extrusion by gentle squashing (Fig. 4a–c). Importantly, red fluorescence was also detected in sperm extruded from the spermatheca of non-transgenic females mated to transgenic males, indicating the usefulness of the Asß2t-DsRed.T3 transgene for sperm marking in A. ludens (Fig. 5d).

Epifluorescent and brightfield images of A. ludens males transformed with pB[XL-EGFP/Asß2tub-DsRed.T3], transformed and non-transformed testes and seminal material from spermathecae dissected from non-transgenic females mated to non-transgenic or transgenic males. Transgenic males (a) under brightfield (left), EGFP epifluorescence showing vector marker expression in the thorax (middle), and DsRed epifluorescence from testes in the posterior abdomen (right); b testes from a transgenic adult male (upper) and a non-transgenic male (lower) under brightfield (left) and DsRed epifluorescence (right) showing fluorescence limited to the transgenic testes; c a gently squashed testis from a transgenic male showing extruded sperm bundles under brightfield (left) and DsRed epifluorescence (right); d a gently squashed spermatheca from a non-transgenic female mated to a non-transgenic male showing extruded sperm under brightfield optics (left), the same spermathecae under DsRed epifluorescence showing a lack of fluorescence in the sperm (middle), and a spermatheca from a non-transgenic female mated to a transgenic male showing extruded fluorescent sperm under DsRed epifluorescence (right)

Brightfield (a) and EGFP epifluorescence (b) of a male (left) and a female (right) pB[XL-EGFP/Asß2tub-DsRed.T3] transgenic showing fluorescence specific to the male, the thoracic flight muscles in particular (with abdominal autofluorescence observed in females)
Discussion
We have genetically transformed the Mexican fruit fly, A. ludens, with two piggyBac transposon vectors and a D. melanogaster hsp70-regulated transposase helper at a reasonably high efficiency, at frequencies similar to, though somewhat higher than initially reported for this species (Condon et al. 2007b). While frequencies for both studies were based on conservative estimates, the first experiments tested different types of vectors having a C. capitata hsp70-regulated transposase helper construct within the vector plasmid (Condon et al. 2007b). In the present study two vectors were introduced which have the potential to improve functional genetic analysis in A. ludens, as well as practical application for control of the pest populations. First, a vector that allows post-integration transgene stabilization, that was first tested in D. melanogaster (Handler et al. 2004), was introduced with subsequent stabilization. The mexfly study differed in that deletion of the 3′ terminal piggyBac sequence by its re-mobilization was catalyzed by an injected piggyBac helper plasmid, and not by mating to a piggyBac jumpstarter strain (having a genomic source of transposase). This has important implications owing to the lack of, or inability to create jumpstarters in many species, and the relative ease of plasmid injection into embryos heterozygous for the vector. At least one less generation is needed to promote re-mobilization, and a subsequent generation to remove the transposase-containing chromosome is not required.
Stabilized vectors expressing only green fluorescence, indicating the loss of the red fluorescent marker and the 3′ terminal sequence from the original vector, were observed at a relatively high frequency since all mating groups yielded progeny expressing only EGFP. Some progeny also expressed only DsRed, which might be explained by the re-mobilized marker transposing to a different chromosome, and segregating from EGFP in the original insertion.
The original post-integration transgene stabilization experiments in Drosophila, that have been duplicated here for the mexfly, showed that loss of a vector terminal sequence eliminates the possibility for subsequent vector re-mobilization by an intended or unintended source of transposase (Handler et al. 2004). This obviates serious programmatic and ecological concerns for transgene instability, which would initially result in loss of desired transgene phenotypes, and then potentially, horizontal transmission of the transgene into unintended host species. The first mexfly transformation study also stabilized vectors, but by re-mobilizing both terminal sequences resulting in transposon-free transgene insertions (Condon et al. 2007b; Dafa’alla et al. 2006). However, these stabilized transgenes are more difficult to achieve, require another marker gene, and the practical need for post-integration deletion of both termini is not compelling. The primary potential limitation of the single terminus deletion approach, that being the fortuitous restoration of a mobilizeable vector, would be extremely unlikely, has never been experimentally demonstrated, and its potential is easily assessed by sequencing the genomic insertion site sequences.
Previous to stabilization, three transgenic lines were subject to several ‘quality’ tests relative to the wild Chiapas host strain, that assessed biological fitness, flight ability and sexual competitiveness. The viability of these strains at all stages and flight ability, indicating overall adult fitness, was lower relative to Chiapas, though typically not by more than a 10–12% differential. This would suggest that a greater number of flies at at least this percentage would have to be reared for an equivalent number of comparably fit flies to be available for release. Diminished fitness in transgenics is not unexpected, since the random nature of transposon-vector insertions often results in mutations having negative effects. This has been borne out by detailed comparisons of discrete transgenic lines from several species, to their non-transgenic parental strain (Catteruccia et al. 2003; Irvin et al. 2004). However, not all transgenic lines have diminished fitness, such as those from Cochliomyia homonivorax (Allen et al. 2004), and probably only require the screening of additional strains having different genomic insertion sites. The eventual development of more optimal target site strains for transgene insertion, that also allow subsequent vector stabilization, should obviate concerns for variable fitness (Horn and Handler 2005; Schetelig et al. 2009).
Arguably, however, the third parameter tested, sexual competitiveness, is likely of much greater importance since strains having a diminished male mating ability may have to be released at a many-fold higher number to compensate. For this parameter, the 11M3 transgenic strain appeared to be equivalent to Chiapas in mating competitiveness, with the other two being less competitive on average, but not at a significant level. Thus, for these transgenic strains, it could be anticipated that for optimal releases, only a minimal increase in numbers would be required for high frequencies of sterile matings (though nevertheless, at at least several-fold higher numbers than the field population).
The second vector inserted into the mexfly contained a DsRed fluorescent marker driven by the spermatocyte-specific ß2-tubulin promoter within the vector pB[XL-PUbEGFP/Asß2t-DsRed.T3] (Zimowska et al. 2009). This marker was originally shown to express in a testis- and sperm-specific manner in A. suspensa, the species from which the ß2-tubulin promoter was derived. Efficient heterologous function for the promoter was not unexpected given the relatively high similarity of sequences 5′ to the transcription initiation site in both species, though a conspicuous 59 bp insertion exists in the mexfly 5′UTR. Testis fluorescence could be detected through the adult abdominal cuticle, and was found to be specific to sperm bundles within the testes. Importantly, red fluorescent sperm were clearly identified when extruded from the spermatheca of non-transgenic females mated to transgenic males. This supports the applicability of the Asß2t-DsRed.T3 transgene for sperm marking in A. ludens, that has the potential use of unequivocally determining whether females trapped in the field have mated with released sterile males.
One of the transgenic lines from the pB[XL-EGFP/Asß2t-DsRed.T3] transformation expressed EGFP only in males, suggesting that it is a Y-linked insertion, yet red fluorescence from the testes was not observed. Conceivably the highly heterochromatic Y chromosome has differentially suppressed the transgene promoters, allowing detection of the EGFP vector marker, but not the ß2-tubulin-regulated DsRed. This possibility was, indeed, tested by re-mobilization of the vector to a new autosomal insertion site that is less suppressive resulting in expression of both the EGFP and the DsRed markers. Nevertheless, Y-linked fluorescence does provide the possibility for male-specific selection, especially if embryonic expression occurs (or is detectable) for fluorescence-based embryonic sorting. A Y-linked insertion of a fluorescent protein-marked vector has been reported for the medfly, Ceratitis capitata (Condon et al. 2007a) and here we report the first Y-linked vector insertion in A. ludens.
In summary, in this study the germ-line of the Mexican fruit fly has been efficiently transformed with two piggyBac-transposon vectors using a non-autonomous transposase helper. One vector allowed post-integration stabilization of transgene sequences that was verified by PCR and sequence analysis. A second vector carried a sperm-specific marker gene regulated by a previously isolated ß2-tubulin promoter from the caribfly, A. suspensa. One of these transgenic lines is male-specific for the transgene marker, suggesting that the vector insertion is Y-linked, and thus providing a possible means for fluorescent-based sexing. As with other tephritid species, this highly significant pest species can now be subject to further manipulation and study using efficient transposon-based transformation methods.
References
Allen ML, Berkebile DR, Skoda SR (2004) Postlarval fitness of transgenic strains of Cochliomyia hominivorax (Diptera: Calliphoridae). J Econ Entomol 97:1181–1185
Alphey L (2002) Re-engineering the sterile insect technique. Insect Biochem Mol Biol 32:1243–1247
Bevis BJ, Glick BS (2002) Rapidly maturing variants of the Discosoma red fluorescent protein (DsRed). Nat Biotechnol 20:83–87
Catteruccia F, Godfray HC, Crisanti A (2003) Impact of genetic manipulation on the fitness of Anopheles stephensi mosquitoes. Science 299:1225–1227
Condon KC, Condon GC, Dafa’alla TH, Fu G, Phillips CE, Jin L, Gong P, Alphey L (2007a) Genetic sexing through the use of Y-linked transgenes. Insect Biochem Mol Biol 37:1168–1176
Condon KC, Condon GC, Dafa’alla TH, Forrester OT, Phillips CE, Scaife S, Alphey L (2007b) Germ-line transformation of the Mexican fruit fly. Insect Mol Biol 16:573–580
Dafa’alla TH, Condon GC, Condon KC, Phillips CE, Morrison NI, Jin L, Epton MJ, Fu G, Alphey L (2006) Transposon-free insertions for insect genetic engineering. Nat Biotechnol 24:820–821
FAO/IAEA/USDA (2003) Manual for Product Quality Control and Shipping Procedures for Sterile Mass-Reared Tephritid Fruit Flies Version 5.0. [IAEA] (International Atomic Energy Agency)
Gutiérrez-Ruelas JM, Santiago-Martínez G (2008) Situación actual de la campaña nacional contra moscas de la fruta en México. In: Montoya-Gerardo PJ, Díaz-Fleischer F, Breceda S (eds) Proceedings: 7th meeting of the working group on fruit flies of the Western Hemisphere. Mazatlan, Sinaloa, México, pp 11–13
Handler AM (2002) Prospects for using genetic transformation for improved SIT and new—biocontrol methods. Genetica 116:137–149
Handler AM (2004) Understanding and improving transgene stability and expression in insects for SIT and conditional lethal release programs. Insect Biochem Mol Biol 34:121–130
Handler AM, Harrell RA (1999) Germline transformation of Drosophila melanogaster with the piggyBac transposon vector. Insect Mol Biol 8:449–458
Handler AM, Harrell RA (2001a) Polyubiquitin-regulated DsRed marker for transgenic insects. Biotechniques 31:820–828
Handler AM, Harrell RA (2001b) Transformation of the Caribbean fruit fly, Anastrepha suspensa, with a piggyBac transposon vector marked with polyubiquitin-regulated GFP. Insect Biochem Mol Biol 31:199–205
Handler AM, McCombs SD, Fraser MJ, Saul SH (1998) The lepidopteran transposon vector, piggyBac, mediates germ-line transformation in the Mediterranean fruit fly. Proc Natl Acad Sci USA 95:7520–7525
Handler AM, Zimowska GJ, Horn C (2004) Post-integration stabilization of a transposon vector by terminal sequence deletion in Drosophila melanogaster. Nat Biotechnol 22:1150–1154
Horn C, Handler AM (2005) Site-specific genomic targeting in Drosophila. Proc Natl Acad Sci USA 102:12483–12488
Irvin N, Hoddle MS, O’Brochta DA, Carey B, Atkinson PW (2004) Assessing fitness costs for transgenic Aedes aegypti expressing the GFP marker and transposase genes. Proc Natl Acad Sci USA 101:891–896
Knipling EF (1955) Possibilities of insect control or eradication through the use of sexually sterile males. J Econ Entomol 48:459–462
McCombs SD, Lee SG, Saul SH (1993) Translocation-based genetic sexing system to enhance the sterile insect technique against the melon fly (Diptera: Tephritidae). Ann Entomol Soc Am 86:651–654
McInnis DO, Lance DR, Jackson CG (1996) Behavioral resistance to the sterile insect technique by Mediterranean fruit fly (Diptera: Tephritidae) in Hawaii. Ann Entomol Soc Am 89:739–744
Orozco-Dávila D, Hernández R, Meza S, Domínguez J (2007) Sexual competitiveness and compatibility between mass-reared sterile flies and wild populations of Anastrepha ludens (Diptera: Tephritidae) from different regions in Mexico. Florida Entomol 90:19–26
Robinson AS, Franz G, Fisher K (1999) Genetic sexing strains in the medfly, Ceratitis capitata: development, mass rearing and field application. Trends Entomol 2:81–104
Robinson AS, Franz G, Atkinson PW (2004) Insect transgenesis and its potential role in agriculture and human health. Insect Biochem Mol Biol 34:113–120
Rull Gabayet JA, Reyes Flores J, Enkerlin W (1996) The Mexican national fruit fly eradication campaign: largest fruit fly industrial complex in the world. In: McPheron BA, Steck GJ (eds) Fruit fly pests: a world assessment of their biology and management. St. Lucie Press, Delray Beach, pp 561–563
Schetelig MF, Scolari F, Handler AM, Kittelmann S, Gasperi G, Wimmer EA (2009) Site-specific recombination for the modification of transgenic strains of the Mediterranean fruit fly Ceratitis capitata. Proc Natl Acad Sci USA 106:18171–18176
Zar JH (1999) Biostatistical analysis, 4th edn. Prentice Hall, Englewood Cliffs
Zepeda-Cisneros CS, Meza JS, Gálvez S, Ibañez J, Robinson AS (2009) Inheritance and linkage studies on eye color mutations in Anastrepha ludens (Diptera: Tephritidae). Ann Entomol Soc Am 103:96–99
Zimowska GJ, Nirmala X, Handler AM (2009) The beta2-tubulin gene from three tephritid fruit fly species and use of its promoter for sperm marking. Insect Biochem Mol Biol 39:508–515
Acknowledgments
Grateful appreciation is extended to the International Atomic Energy Agency (IAEA) for funding though the Technical Co-operation Project MEX/5/027. Additional support was provided by the USDA-NIFA-Agriculture and Food Research Initiative (AMH) and the Campaña Moscas de la Fruta (Mexican Fruit Fly Campaign) DGSV-SAGARPA.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10709_2010_9484_MOESM2_ESM.doc
Supplementary Figure 1 PCR product sequences for the 5’ and 3’ insertion sites for the pB[L1-EGFP-L2-DsRed-R1] vector in genomic DNA from the 11M3 green/red strain and the L1-EGFP sequence stabilized by post-integration deletion of the L2-DsRed-R1 sub-vector in the 11M3 green strain. PCR products were extracted from indicated lanes (see Fig. 2 and Table 3) and sequenced from subclones using either M13 or internal primers. (A) shows the same sequence for the 5’ insertion site for both the 11M3 green/red (lane 1) and green (lane 5) strains using the 534F and 122R primers; (B) shows the 3’ insertion site sequence for the 11M3 green/red strain using primers 144F and 515R; and (C) shows the 3’ insertion site sequence for the 11M3 green strain (lane 9) using primers 387F and 515R after deletion of the L2-DsRed-R1 sub-vector. Primers sites are indicated by arrows below or above, the TTAA duplicated insertion site is double-underlined, piggyBac terminal sequences are in bold (A, B), and EGFP/SV40 sequences are single-underlined (C)
Rights and permissions
About this article
Cite this article
Meza, J.S., Nirmala, X., Zimowska, G.J. et al. Development of transgenic strains for the biological control of the Mexican fruit fly, Anastrepha ludens . Genetica 139, 53–62 (2011). https://doi.org/10.1007/s10709-010-9484-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-010-9484-6