
Abstract
Insect pest control programs incorporating the sterile insect technique (SIT) rely on the mass production and release of sterilized insects to reduce the wild-type population through infertile matings. Most effective programs release only males to avoid any crop damage caused by female fruit flies or transmission of disease by female mosquitoes. Therefore, the females have to be eliminated, preferably in an early developmental stage, during mass rearing. Different systems and techniques have been created for the sex separation of a few insect species. One of these is the transgenic sex-specific fluorescent protein marking of the insects with automated fluorescent-based sorting of the individuals to achieve sex separation. Here we describe the Y-linked integration of fluorescent markers driven by the widely active Drosophila melanogaster polyubiquitin promoter in the Caribfly, Anastrepha suspensa. Four strains with Y-linked integrations were established with one line expressing the DsRed fluorescent protein marker during embryogenesis. This line now has the possibility for use with automated sex separation in rearing, and the same transgene markers could be used in other insects for similar applications.


Graphical Abstract
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Integrated pest management (IPM) programs have been used to efficiently control pest species around the world. IPM is a sustainable approach to manage pests by combining biological, cultural, physical, and chemical tools in a way that minimizes economic, health, and environmental risks [1]. An important element of many IPM programs is the sterile insect technique (SIT), which is an environmentally friendly and species-specific program for efficient population control. SIT is an area-wide applied process in which reproductively sterile males are released at overflooding ratios into a wild population of the same species, resulting in preferential mating with wild females in the field that are rendered nonreproductive [2]. For the most effective SIT programs, the species must be mass-reared, sexed early in development (separation of males and females), marked for monitoring, and sterilized by irradiation before release into affected areas. In particular, the production of a male-only population is highly important for large-scale SIT programs because this is most efficient and cost-effective for fruit fly programs [3, 4] and a prerequisite for mosquito programs where adult females are vectors of disease [5, 6]. For most biologically based control release programs, it is highly desirable to have females eliminated early in development to avoid female larval feeding in the mass rearing process [7]. The most commonly used methods are physical, genetic, and transgenic techniques for sex separation.
Physical sexing includes manual sorting using external morphological differences or automated machine sorting based on sex-specific size or color variation. Manual sorting can be labor intensive, but it has been used for sex separation of the tsetse fly, Glossina austeni. New knowledge of timing differences between male and female adult emergence has improved sexing and eliminated the need for laborious hand sorting [8, 9]. In the melon fly, Bactrocera cucurbitae [10], the mexfly, Anastrepha ludens (J.S. Meza, personal comm.), and the medfly, Ceratitis capitata [11], strains with sex-specifically colored pupae have been developed. For the melon fly strain, pupae were sexed with high-speed photoelectric sorting machines [12], which could be applied to the other species as well. All physical sorting techniques have the disadvantage that both sexes must be reared at least through larval stages (and typically to the pupal stage), which increases production costs in mass rearing.
Another option for sexing is the creation of genetic sexing strains (GSS) by classical genetic manipulations. In the Mediterranean fruit fly, a GSS has been developed and refined throughout the last 20 years. It is based on two separate components: (i) a temperature-sensitive lethal (tsl) mutation that is maintained in both sexes as homozygous alleles, and (ii) a Y chromosome translocation that carries the wild-type allele (tsl +) for the mutation in only the males. In this way, GSS mutant females are eliminated early in development at elevated temperatures, while males survive owing to the Y-linked presence of the wild-type allele [7]. The medfly GSS is currently used in mass rearing to produce up to 4 billion flies per week. The difficulty of transferring such a system to other insects is due to the unpredictable process of isolating tsl mutations and the induction of translocations and stabilizing inversions in species that are not genetically well-characterized [7].
In mosquitoes, similar GSSs have been developed since the 1970s based on dominant temperature sensitive (DTS) mutations [13] or insecticide resistance to dieldrin [5, 6]. As with the tsl GSS, the mutations could be homozygous in both sexes, while only males carry the rescuing wild-type (WT) allele on the Y chromosome through an induced translocation. However, for dieldrin-dependent GSSs, relatively high semisterility of the males has been problematic for expanding the production capacity, and the waste management of dieldrin-containing solutions and diets in large-scale production is of concern.
Transgenic sexing systems based on lethality systems that are conditionally-repressed by tetracycline were first developed and tested in D. melanogaster [14, 15] and then transferred to medfly and Bactrocera oleae [16, 17]. These systems are able to kill a high percentage of females when the lethality system is combined with alternative, sex-specific splicing of the medfly transformer intron. However, for these systems, the majority of the female lethality occurs at late larval or early pupal stages, which increases mass rearing costs due to the feeding of female larvae. Recently, transgenic embryonic sexing strains (TESSs) were developed in Anastrepha suspensa and C. capitata [18, 19]. Both systems are also based on a tetracycline repression system, but they induce lethality during embryogenesis by the use of embryo-specific promoters and proapoptotic lethal effectors. For both species, several TESSs were generated with 100 % early lethality as confirmed in large-scale tests [19].
Another transgenic technology, which could be transferred to other insects, is male-specific marking by fluorescent proteins. Such strains have been developed in mosquitoes and fruit flies by inserting a transgene carrying a fluorescent protein under the control of a testis-specific or constitutive promoters [20–23]. Automated fluorescence sorters (COPAS, Union Biometrica) could then be used to separate male and female larvae. In mosquitoes, this approach was successfully used to create a male-only population for Anopheles gambiae [24]. Nevertheless, female larval rearing is still necessary; for fruit flies, late larval stages may be too large for the automated sorting machine. Fluorescent marking of physically smaller embryos or early larval stages would improve the throughput of the separation system and avoid larval feeding during mass production. To address these limitations, we describe the creation of male-specifically expressed Y-linked transgene integrations in A. suspensa and the evaluation of early male-specific fluorescence for separating males and females.
2 Results
2.1 Random Integration of Transgenes in A. suspensa
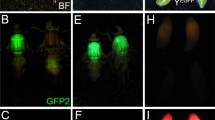
To generate male-specific fluorescent marked strains, the markers need either to be linked to the Y chromosome or be under the influence of a male-specific promoter/enhancer. Because there is no site-specific targeting available for A. suspensa to insert markers directly onto the Y chromosome or other preferred male-specific chromosomal loci, three piggyBac vectors (423_attP_PUbEGFP, 437_attP_PUbDsRed, and 443_attP_PUbEGFP) were integrated into the fly genome by germline transformation. The vectors were integrated into an A. suspensa WT strain, establishing 5 to 10 independent lines each by screening for epifluorescence of the PUb-EGFP or PUb-DsRed.T3 marker. Randomly, 4 out of 20 independent lines expressed the fluorescent markers male-specifically (437_M5A, 437_M7A, 423_M10B, 443_M7m5; Fig. 1). Transgenic males from these lines were backcrossed to WT females to determine Y chromosome transgene linkage of their offspring by epifluorescence screening. All male progeny from the strains 437_M5A, 437_M7A, 423_M10B, and 443_M7m5 expressed the respective fluorescent marker, whereas none of the females exhibited any marker expression. This backcross was repeated with the selected males crossed to WT females and resulted again in 100 % fluorescently marked males. This indicated Y-linkage of the fluorescent marker because an autosomal insertion would not yield expression in all males.


Male-specifically marked A. suspensa strains. All lines show male-specific expression of the PUbDsRed.T3 (a and b) or the PUbnlsEGFP marker (c and d). Males and females from each line were observed under brightfield conditions (left panel) and epifluorescence microscopy with the respective filter sets, YFP or TxRed (right panel)
2.2 Molecular Characterization
For each transgene, the integration site flanking sequences were isolated by thermal asymmetric interlaced (TAIL) polymerase chain reaction (PCR), confirming that all were canonical piggyBac integrations into genomic TTAA sites. For 437_M5A, the genomic flanking sequences were identified as a microsatellite locus similar to a previously described A. suspensa 1-5E microsatellite clone by the BLASTN algorithm using the nr database at NCBI [25], whereas the integration site of 443_M7m5 had similarities to an A. suspensa mariner transposase pseudogene (accession number U04466). Interestingly, two of the four integrations, 437_M7A and 423_M10B, occurred independently in the same intron of a gene homologous to the male-specifically expressed gene, CG14830, in D. melanogaster [26] and a testes developmental protein, nyd-sp29, in Ae. aegypti (Fig. 2a). The Y-linked integration of the transgene in the strains 437_M5A and 443_M7m5 was further verified by PCR to the isolated flanking regions to female and male genomic DNA (Fig. 2b).


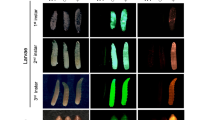
Genomic verification of integrations and embryonic fluorescence. a Genomic structure of As-CG14830 (A. suspensa cognate of D. melanogaster CG14830) indicating independent piggyBac integration events in the lines 423_M10B and 437_M7A. Base pair numbers indicate the isolated region of the As-CG14830 gene (see also Supplementary data). b PCR on male (m) and female (f) genomic DNA of 437_M5A and 443_M12m1 targeting the tTA and EGFP gene, respectively. c Embryos from 437_M5A males and females under brightfield conditions (left panel) and epifluorescence microscopy with the TxRed filter set (right panel)
2.3 Sex Separation of Fluorescent Embryos
To perform automated embryonic sorting and avoid larval rearing, detection of embryonic fluorescence is essential. Therefore, embryos and larvae from all Y-linked strains were examined for the earliest visible fluorescence. In 437_M7A, 423_M10B, and 443_M7m5, expression of DsRed or EGFP was not detected until the third instar larval stage. The only strain expressing the DsRed by late embryogenesis (55–64 h after egg laying) and the first larval instar was 437_M5A. A total of 200 embryos from this strain that did (97) or did not (103) express DsRed were then manually selected and maintained on two different larval diet plates. From the DsRed-expressing embryos, 69 survived as male-only adults, whereas 72 adult females enclosed from the nonexpressing embryos. This demonstrated that progeny from the line 437_M5A could be separated during embryogenesis to create a male-only population (Fig. 2c).
3 Discussion
Here we describe the generation of the first Y-linked markers for use in embryonic sex separation in the tephritid pest, A. suspensa. Four transgenic strains were established with one expressing a fluorescent protein during embryogenesis. Integrations in A. suspensa with male-specific expression patterns thus occurred in 20 % of independent strains generated in this experiment, although this relatively high frequency is likely to differ for other insects and with the use of other transgene vectors. The three different constructs tested in A. suspensa were distinct except for piggyBac vector sequences and a 220 bp attP landing site, so it is unlikely that transgene vector structure was responsible for the generation of the male-specifically expressing lines. Interestingly, two vectors integrated independently into the same intron of a gene having homology to a testes developmental protein gene in Ae. aegypti, although testis-specific fluorescence was not detected. At present, it remains to be determined whether male-specific enhancers effect the sex-specific expression of the fluorescent marker.
It is more likely that Y-linkage for the transgene is primarily responsible for the male-specific marker expression. Although a molecular determination for Y-linkage has yet to be determined for either integration (due to a lack of Y-specific sequence data), Y insertions are supported by backcrosses of fluorescent-marked males to WT females that resulted in only fluorescent male progeny. Thus, for the first time, a Y-linked embryonic fluorescent expression line, 437_M5A, has been created that can be used to separate male from female embryos to generate a male-only population. In addition, the #437 transgene has an attP landing site that can be used to integrate new transgenes specifically on the Y chromosome, providing a reliable mechanism for future male-specific modification of these strains.
The marking of males by fluorescent proteins had been previously achieved in several other insects [20, 22, 27], but fluorescence did not appear before late larval stages, with the exception of one system for the Mediterranean fruit fly [21]. Because feeding larvae increases production costs in large-scale rearing, developing embryonic sexing systems remained a high priority. The strategy of randomly integrating markers driven by the polyubiquitin (PUb) promoter onto the Y chromosome is an alternative to physical, genetic, or other transgenic sexing techniques. Such markers have the advantage that they can be easily transferred to new species, whereas other highly efficient embryonic transgenic sexing systems need more adaptation to the host species [18, 19]. Development of Y-linked markers can be typically achieved in a shorter timeframe than generating classical genetic sexing strains [7], but a limiting factor is the need for separation by automated fluorescence sorting machines. Currently, these machines can sort material only up to a certain size, which can be problematic for insects having large embryos or first instar larvae. Recently, sorting fluorescently labeled larvae of An. gambiae by the automated COPAS system has been evaluated [24]. This was highly accurate for both transgene heterozygotes and homozygotes; however, a high-throughput evaluation over several days has yet to be conducted. Presently, the calculated, sortable numbers are not sufficient for SIT programs using 100 millions of males weekly. In the future, further improvements on sorting machines could eliminate this bottleneck, allowing high-throughput separation to be possible for many insect species.
4 Materials and Methods
Insect rearing. An inbred wild-type colony of Anastrepha suspensa (Homestead, Florida) was maintained at 25 °C and reared under standard laboratory conditions [28, 29]. All embryonic, larval, and pupal stages of A. suspensa were reared at 27 °C and 60 % humidity on a 12 h light:12 h dark cycle.
Cloning. The vector #423 (pXLII_PUbEGFP_f_attP235_SV40-slamA-AstraIntron-slam_hs43-TRE) was created by ligating a FseI/AscI cut 5.6 kb attP235_SV40-slamA-AstraI-slam_hs43-TRE fragment into FseI/AscI cut vector #1419 [30]. The vector #437 (pXLII_attP_PUbDsRedT3_Ccvas-tTA) was generated by ligating the AscI fragment Ccvas-tTA from M493 to the AscI cut vector #1425 [30]. To create M493, the Ccvas promoter was SmaI/XbaI cut from M429 and ligated into #1215 [31].
The vector 443 (pBXLII_PUbEGFP_TREhs43-CctraI-Alhid Ala2 _loxN-3xP3-FRT-AmCyan_lox2272_loxP_attP235) was described previously [19].
Germline transformation. Germline transformation experiments were performed by microinjection of the piggyBac constructs #423 or #437 (500 ng/μl) together with the phsp-pBac transposase helper plasmid (200 ng/μl) into WT A. suspensa embryos as described [32]. G1 offspring were selected by EGFP or DsRed epifluorescence using a Leica MZ FLIII microscope and the YFP (ex: 500/20; em: 535/30) or TxRed (ex: 560/40; em: 610 LP) filter sets.
Independent homozygous strains were established by single pair inbreeding for successive generations with testing by segregation analysis of transformants outcrossed to WT flies. Transgenic A. suspensa lines carrying the #443 piggyBac cassette were generated and described earlier as a lethal effector construct [19].
Isolation of transgene integration flanking site sequences. Flanking sequences of #423, #437, and #443 transgene integrations were isolated by TAIL PCR or inverse PCR. TAIL PCR conditions were as described previously [33]. Oligos used for the isolation of the 5′ piggyBac vector insertion-site flanking sequences (of 423_M10B, 437_M5A, and 443_M7m5) by TAIL PCR were the degenerate primer AD3 (AGWGNAGWANCAWAGG) and the specific primers L1_P882 (CATTTTGACTCACGCGGTCGTTATAGTTC), L2_P883 (CAGTGACACTTACCGCATTGACAAGCA), and L3_P884 (CGACTGAGATGTCCTAAATGCACAG). Oligos for the 3′ piggyBac flanking sequence (of 437_M5A and 443_M7m5) were the degenerate primer AD3 (AGWGNAGWANCAWAGG) and the specific primers R1_P885 (ACCTCGATATACAGACCGATAAAACACATGC), R2_P886 (GTCAATTTTACGCATGATTATCTTTAACGT), and R3_P887 (CGTACGTCACAATATGATTATCTTTCTAGG). The PCR conditions and the generation of DNA pools for inverse PCR are described in Schetelig and Handler (2012a). The 3′ flanking sequences of 423_M10B and 437_M5A were isolated by inverse PCR using XhoI-digested genomic DNA and the oligo pairs P144/P830 (CCTCGATATACAGACCGATAAAACAC/CTTTTATCGAATTCCTGCAGC) and P144/P777 (CCTCGATATACAGACCGATAAAACAC/CCGACATGACACAAGGGGTTG), respectively.
Verification of Y-linked integrations. First, the expression of fluorescent markers in adult flies was used to identify male-specifically marked strains by epifluorescence microscopy. To confirm Y-linked transgene insertions, fluorescent males were subsequently backcrossed to WT A. suspensa females for two generations and the number of fluorescent/nonfluorescent progeny assessed. Epifluorescence was also used to assess embryos and larvae from male-specifically expressing lines for the earliest possible stage of marker detection using the YFP or TxRed filter sets.
Secondly, transgene integrations in the lines 437_M5A and 443_M7m5 were molecularly verified by PCR of genomic DNA (PCR conditions: 2 min at 95 °C; 30 cycles of 20 s at 94 °C, 30 s at 59 °C, 20 s at 72 °C). The oligo pairs P756/P757 (GCTGCTTAATGAGGTCGGAATCG/TGGTGCCTATCTAACATCTCAATGG), binding to the tTA gene of the transgene #437, and P913/P914 (CAGAACACCCCCATCGGCGACGGC/TACTTGTACAGCTCGTCCATG), binding to the EGFP marker of #443, were then used on male and female genomic DNA.
References
Klassen W, Curtis CF (2005) History of the Sterile Insect Technique. In: Dyck VA, Hendrichs J, Robinson AS (eds) Sterile insect technique principles and practice in area-wide integrated pest management. Springer, Dordrecht, pp 3–36
Knipling EF (1955) Possibilities of insect control or eradication through the use of sexually sterile males. J Econ Entomol 48:459–462
Mcinnis DO, Tam S, Grace C et al (1994) Population suppression and sterility rates induced by variable sex ratio, sterile insect releases of Ceratitis capitata (Diptera: Tephritidae) in Hawaii. Ann Entomol Soc Am 87:231–240
Rendon P, Mcinnis D, Lance D et al (2004) Medfly (Diptera: Tephritidae) genetic sexing: large-scale field comparison of males-only and bisexual sterile fly releases in Guatemala. J Econ Entomol 97:1547–1553
Baker RH, Sakai RK, Raana K (1981) Genetic sexing for a mosquito sterile-male release. J Hered 72:216–218
Yamada H, Benedict MQ, Malcolm CA et al (2012) Genetic sex separation of the malaria vector, Anopheles arabiensis, by exposing eggs to dieldrin. Malar J 11:208
Franz G (2005) Genetic sexing strains in Mediterranean fruit fly, an example for other species amenable to large-scale rearing for the sterile insect technique. In: Dyck VA, Hendrichs J, Robinson AS (eds) Sterile insect technique—principles and practice in area-wide integrated pest management. Springer, Dordrecht, pp 427–451
Opiyo E, Luger D, Nadel D et al (1999) Automation in tsetse mass-rearing process: preliminary observations with Glossina austeni. In: Proceedings of animal trypanosomosis: vector and disease control using nuclear techniques. Second FAO/IAEA seminar for Africa, Zanzibar, Tanzania. Backhuys Publishers, Leiden, NL, 27 Nov–1 Dec 1995, p 187–192
Opiyo E, Luger D, Robinson AS (2000) New systems for the large-scale production of male tsetse flies (Diptera: Glossinidae). In: Tan KH (ed) Proceedings: area-wide control of fruit flies and other insect pests, and the 5th international symposium on fruit flies of economic importance, 28 May–5 June 1998, Penang, Malaysia. Penerbit Universiti Sains Malaysia, Pulau Pinang, Malaysia, p 337–344
Mcinnis DO, Lim R, Muromoto D et al (2005) Oriental fruit fly: males-only sterile fly releases in Hawaii. In: The 8th exotic fruit fly symposium. Riverside, California, p 34
Rössler Y (1979) The genetics of the Mediterranean fruit fly: a ‘white pupae’ mutant. Ann Entomol Soc Am 72:583–585
Mcinnis D, Leblanc L, Mau R (2007) Melon fly (Diptera: Tephritidae) genetic sexing: all-male sterile fly releases in Hawaii. Proc Hawaiian Entomol Soc 39:105–110
Baker RH, Sakai RK, Saifuddin UT (1978) Genetic sexing technique for a mosquito sterile male release. Nature 274:253–255
Heinrich JC, Scott MJ (2000) A repressible female-specific lethal genetic system for making transgenic insect strains suitable for a sterile-release program. Proc Natl Acad Sci USA 97:8229–8232
Thomas DD, Donnelly CA, Wood RJ et al (2000) Insect population control using a dominant, repressible, lethal genetic system. Science 287:2474–2476
Ant T, Koukidou M, Rempoulakis P et al (2012) Control of the olive fruit fly using genetics-enhanced sterile insect technique. BMC Biol 10:51
Fu G, Condon KC, Epton MJ et al (2007) Female-specific insect lethality engineered using alternative splicing. Nat Biotechnol 25:353–357
Ogaugwu CE, Schetelig MF, Wimmer EA (2013) Transgenic sexing system for Ceratitis capitata (Diptera: Tephritidae) based on female-specific embryonic lethality. Insect Biochem Mol Biol 43:1–8
Schetelig MF, Handler AM (2012) A transgenic embryonic sexing system for Anastrepha suspensa (Diptera: Tephritidae). Insect Biochem Mol Biol 42:790–795
Catteruccia F, Benton JP, Crisanti A (2005) An Anopheles transgenic sexing strain for vector control. Nat Biotechnol 23:1414–1417
Condon KC, Condon GC, Dafa’alla TH et al (2007) Genetic sexing through the use of Y-linked transgenes. Insect Biochem Mol Biol 37:1168–1176
Scolari F, Schetelig MF, Bertin S et al (2008) Fluorescent sperm marking to improve the fight against the pest insect Ceratitis capitata (Wiedemann; Diptera: Tephritidae). N Biotechnol 25:76–84
Smith RC, Walter MF, Hice RH et al (2007) Testis-specific expression of the β2 tubulin promoter of Aedes aegypti and its application as a genetic sex-separation marker. Insect Mol Biol 16:61–71
Marois E, Scali C, Soichot J et al (2012) High-throughput sorting of mosquito larvae for laboratory studies and for future vector control interventions. Malar J 11:302
Fritz A, Schable N (2004) Microsatellite loci from the Caribbean Fruit Fly, Anastrepha suspensa (Diptera: Tephritidae). Mol Ecol 4:443–445
Graveley BR, Brooks AN, Carlson JW et al (2011) The developmental transcriptome of Drosophila melanogaster. Nature 471:473–479
Zimowska GJ, Nirmala X, Handler AM (2009) The beta2-tubulin gene from three tephritid fruit fly species and use of its promoter for sperm marking. Insect Biochem Mol Biol 39:508–515
Roberts DB (1986) Drosophila: a practical approach. IRL Press, Oxford
Saul SH (1982) Rosy-like mutant of the Mediterranean fruit fly, Ceratitis capitata (Diptera: Tephritidae), and its potential for use in a genetic sexing program. Ann Entomol Soc Am 75:480–483
Schetelig MF, Handler AM (2012) Strategy for enhanced transgenic strain development for embryonic conditional lethality in Anastrepha suspensa. Proc Natl Acad Sci USA 109:9348–9353
Schetelig MF, Caceres C, Zacharopoulou A et al (2009) Conditional embryonic lethality to improve the sterile insect technique in Ceratitis capitata (Diptera: Tephritidae). BMC Biology 7:4
Handler AM, Harrell RA (1999) Germline transformation of Drosophila melanogaster with the piggyBac transposon vector. Insect Mol Biol 8:449–457
Liu YG, Whittier RF (1995) Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 25:674–681
Acknowledgments
We thank Shelley Olson for excellent technical assistance. Funding is gratefully acknowledged form the USDA-NIFA-Agriculture and Food Research Initiative (AMH) and the Emmy Noether Program SCHE 1833/1 of the Deutsche Forschungsgemeinschaft (MFS).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Supplementary data
Supplementary data
Sequence of the A. suspensa CG14830 homolog. TTAA piggyBac integration sites of the plasmids 423 (green) and 437 (red) are indicated. Possible exons predicted by comparison to D. melanogaster CG14830 are marked (orange).


Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Schetelig, M.F., Handler, A.M. (2013). Y-Linked Markers for Improved Population Control of the Tephritid Fruit Fly Pest, Anastrepha suspensa . In: Vilcinskas, A. (eds) Yellow Biotechnology II. Advances in Biochemical Engineering/Biotechnology, vol 136. Springer, Berlin, Heidelberg. https://doi.org/10.1007/10_2013_209
Download citation
DOI: https://doi.org/10.1007/10_2013_209
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-39901-5
Online ISBN: 978-3-642-39902-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)