
Abstract
In this study we analysed mitochondrial DNA variation in Penaeus kerathurus prawns collected from seven locations along a transect across the Siculo–Tunisian region in order to verify if any population structuring exists over a limited geographical scale and to delineate the putative transition zone with sufficient accuracy. Partial DNA sequences of COI and 16S genes were analysed. In contrast to the highly conservative 16S gene, the COI sequences exhibited sufficient diversity for population analysis. The COI gene revealed low levels of haplotype and nucleotide diversities. The size of the annual landings of this commercial species suggests large population sizes. Hence, the low genetic diversity detected in this study could indicate a possible reduction in effective population sizes in the past. We detected significant genetic differentiation between eastern and western populations likely due to restricted gene flow across the Siculo–Tunisian boundary. We discuss the different evolutionary forces that may have shaped the genetic variation and suggest that the genetic divide is probably maintained by present-day dispersal limitation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In marine environments, where effective physical barriers are difficult to occur, most species spend part of their life cycle in open waters as free-moving gametes, larvae or adults. The expected pattern is that species with high dispersal capabilities (i.e. planktotrophic larvae and continuous environment) have little genetic structure and high gene flow (Lacson 1992; Ovenden et al. 1992; Russo et al. 1994; Uthicke and Benzie 2003). Nevertheless, broad-scale surveys of genetic variation within some marine species have shown that many are genetically more structured than expected (Avise 2000; Hellberg 1996; Lemaire et al. 2005), because a variety of factors including biological, ecological, physiological, physical and geological factors might contribute to the shaping of the population structure of marine species through space and time. In this regard, population genetics and phylogeographic studies are necessary in order to examine the spatial and temporal scales at which populations are genetically structured and can help us understand how speciation takes place in the open sea.
Between 5.96 and 5.33 Myr ago, isolation of the Mediterranean Sea from the Atlantic Ocean led to the Messinian salinity crisis during which sea-levels dropped considerably, reducing the Mediterranean Sea into hypersaline lakes (Krijgsman et al. 1999). Most of the species comprising the present-day fauna have colonized the Mediterranean due to the opening of Gibraltar strait at the end of Miocene. During the Pleistocene-era, glacial episodes involved cyclical environmental variations with associated marine regressions and transgressions directly affecting population distributions and demographics (Hewitt 1996; Avise 2000). The large climatic fluctuations during the Quaternary have led to the assumption that vicariance is the most likely model of speciation promoting genetic discontinuities across geographical ranges (McMillan and Palumbi 1995; Cunningham and Collins 1998; Benzie 2000). However, other contemporary factors that limit effective genetic dispersal, including oceanographic currents, isolation-by-distance, habitat discontinuities, larval behaviour and local adaptation (Johnson and Black 1995; Benzi and Williams 1997; Palumbi et al. 1997; Schmidt and Rand 1999; Riginos and Nachman 2001), may also play a pivotal role. These factors may be important singly or in combination, and their relative contributions are hard to disentangle. The predominant mechanisms leading to population differentiation are however, not always clear (Palumbi 1994).
Places where genetic transitions occur on a limited geographical scale may provide opportunities to study these mechanisms because one expects to find the various forces that may reduce gene flow. A number of studies have hypothesized the existence of a genetic transition in the Mediterranean around the Siculo–Tunisian (S–T) strait for several species (e.g. bivalves, Quesada et al. 1995; Nikula and Vainola 2003; several fish species, Borsa et al. 1997; sea bass, Bahri-Sfar et al. 2000 and seagrass, Arnaud-Haond et al. 2007). Almost all these authors surveyed populations from very distant localities and did not specifically concentrate their sampling efforts on the regions surrounding the S–T strait. This raises the following question: do the differentiation they depicted between eastern and western Mediterranean populations actually due to the S–T barrier itself? The Mediterranean has sometimes been referred to as a ‘sea of seas’ because of its division into different subbasins, each with its own distinct characteristics, including partially enclosed gyral current systems and likely different ecological conditions. Thus distinct hydrographic and ecological conditions may be sufficient to reduce gene flow even on a relatively small geographic scale within seemingly continuous populations (see for example Waples 1987; Planes et al. 1995; Magoulas et al. 2006). Furthermore, precision in delineating transition zones depends on sampling schemes. For instance, the genetic boundary between northeastern Atlantic and the western Mediterranean populations of the mussel Mytilus galloprovincialis has been suspected to coincide with the Gibraltar strait (Borsa et al. 1997). However, appropriate sampling in the same species showed that the genetic discontinuity actually corresponds in position with the Almeria–Oran oceanographic front some 400 km eastward the strait (Quesada et al. 1995).
The caramote prawn Penaeus (Melicertus) kerathurus (Forskäl 1775) is an ecologically and economically important penaeid species. It is widespread in the Mediterranean and ranges from the south coast of England to Angola in the Eastern Atlantic. The benthic adults inhabit nearshore and offshore waters to a depth of about 80 m and prefer muddy or sandy-mud flats. In summer, adults migrate to reproduce in coastal areas and spawn in offshore waters. After a planktonic larval stage (about 4 weeks) post-larvae move into shallow waters, where they enter the juvenile stage until they reach 5–8 cm in length and then join the adult population (Garcia and Le Reste 1981). This species is extensively fished; the annual global capture production is around 6 million kg (FAO 2006). Despite its economic and ecological importance, little is known on the different aspects of P. kerathurus biology or genetics and almost no data are available on the stock structure (see Benzie 2000 for review). A recent study based on allozymes showed low genetic diversity and suggested population structuring over a relatively short geographical scale (Zitari-Chatti et al. 2008).
The present study sampled P. kerathurus from seven localities along a transect (from Naples, Italy to Djerba, Tunisia) of about 1,500 km of the species range in order to obtain a fine-scale spatial coverage of the S–T region. We analysed sequence variation of two mtDNA genes in order to verify if any population structuring exists over a limited geographical scale and to localize the putative transition zone with sufficient accuracy.
Materials and methods
Sampling
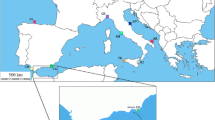
Eighty-four individuals were collected from inshore sites at seven locations ranging from North to South Tunisia and one location in Italy (Naples) (Fig. 1). These samples belong to two regions: the western region represented by Naples, Tabarka and Kalatlandalos locations and the eastern region represented by the four remaining sample locations. All samples were obtained from commercial fleet. Ten or more individuals were analysed per site (Table 1). Muscle tissue was extracted from fresh shrimp and immediately frozen at −80°C until processed. All individuals, except the Naples specimens, were surveyed in a previous allozyme study (Zitari-Chatti et al. 2008).

Geographic location of the seven samples of Penaeus kerathurus. For the full names of locations see Table 1
DNA extraction, amplification and sequencing
Total genomic DNA was extracted using the Nucleospin kit following the instructions of the supplier (Clontech, Maountain view, USA). The concentration of extracted DNA was spectrophotometrically estimated. The lowest DNA concentration was estimated to be about 30 ng μl−1. PCR amplification of a ~562 bp fragment of the 16S rRNA gene (16S) and of a ~374 bp fragment of the mitochondrial cytochrome oxidase c subunit I gene (COI) were carried out using respectively, the primers reported in Simon et al. (1994), 16Sar (5′-CGCCTGTTTATCAAAAACAT-3′) and 16Sbr (5′-CCGGTCTGAACTCAGATCACGT-3′), and our primers derived from P. kerathurus partial COI gene sequence (Lavery et al. 2004) COI1 (5′-GGGTTCGTAGTCTGAGCACACC-3′) and COI2 (5′-TTAGGGTTAAGGGTTAAGCCGG-3′). The reaction volume (50 μl) for 16S contained 3 μl of undiluted DNA extract, 2.5 mM MgCl2, 0.12 μM from each primer, 400 μM of dNTPs, 1 U of Taq polymerase and ddH2O. The PCR mix and volume for COI were similar except that 4 mM of MgCl2, 0.16 μM from each primer, 480 μM of dNTPs were used. The cycling profile for the two genes was one step at 95°C for 3 min followed by 35 cycles at 94°C for 30 s, 50°C for 30 s, and 72°C for 30 s, and a final 5 min extension step at 72°C. The size and the quality of PCR products were visualised on 1% agarose gels. PCR products were cleaned with the GeneElute™ PCR DNA purification kit (Sigma, St Louis, USA) and directly sequenced using the BigDye™ terminator cycle sequencing chemistry, following the manufacturer’s protocol (Applied Biosystems, Foster City, USA). The sequences were recorded with an ABI 97 3100 automated sequencer (Perkin-Elmer, Waltham, USA).
Data analysis
All sequences were aligned using the multiple-alignment programme clustal w (Thompson et al. 1994) with adjustments made by eye. The data sets for both COI and 16S genes were analysed independently. The alignment of COI sequences was confirmed by translating the aligned DNA sequences into amino acid. Identification of the COI and 16S fragments was confirmed by comparing our sequences with those for P. kerathurus published by Lavery et al. (2004). Data analyses were performed using arlequin 3.0 (Excoffier et al. 2005) except where noted. The nucleotide composition, number of transitions/transversions, number of haplotypes and haplotype diversity (h) and nucleotide diversity (π) values (Nei 1987) were calculated. The total number of nucleotide differences and percent sequence divergence values were calculated between each pair of haplotypes.
To assess the geographic structuring of collections, we calculated the pairwise genetic distances (F ST) and their significance by performing 10,100 permutations among the individuals between populations. We also performed a hierarchical analysis of molecular variance (AMOVA; Excoffier et al. 1992) pooling the populations in western and eastern Mediterranean groups. The statistical significances of the F ST values were tested by executing 16,002 permutations. The sequential Bonferroni test (Rice 1989) was used to correct for multiple tests.
To determine whether there was a relationship between genetic distance and geographical distance, we used the Mantel test (in genetix version 4.03; Belkhir et al. 2001) on matrices of standardized pairwise F ST values [F ST/(1 − F ST)] and pairwise geographical distances. The geographical distance between each pair of populations was estimated by measuring the linear map distance.
Relationships among haplotypes were analysed in a minimum-spanning network estimated with minspnet programme implemented in arlequin, using the total number of nucleotide differences. This method provides a 95% plausible set for all sequences type linkages within an unrooted tree.
Results
Gene and haplotype diversities
Nucleotide sequences of the COI and 16S fragments were determined for 84 P. kerathurus individuals (Table 1). Final truncated sequences analysed were 296 and 454 bp for COI and 16S, respectively. The nucleotide composition of the COI sequences averaged 23% C, 33% T, 27% A and 17% G. There were five transitions and one transversion and no indels. A total of seven different haplotypes was found (GenBank Accession numbers EU430763-EU430769), their respective frequencies in each population are described in Fig. 2. The seven haplotypes differed from one another by 1–4 mutations (mean = 2.19, SD = 0.90), and had pairwise sequence divergence values ranging from 0.34 to 1.37%. All the mutations resulted in synonymous substitutions. The overall π–value was 0.0022 (SD = 0.0019) and the within population π–values ranged from 0.0005 to 0.0033 (Table 1). All populations showed low values of haplotype diversity (mean 0.546 ± 0.047) and low values of nucleotide diversity (mean 0.0022 ± 0.0019) (Table 1).

Mitochondrial COI haplotype frequencies in the seven samples of P. kerathurus. Less frequent haplotypes present in eastern (h3, h4 and h5) and western (h6 and h7) samples were combined, respectively under the ‘eastern pool’ and ‘western pool’
For 16S sequences, base composition averaged 12% C, 34% T, 34% A and 20% G. There were only three haplotypes differing by 1–2 mutations (GenBank Accession numbers EU430760-EU430762), one haplotype (haplotype no. 1 present in 96% of the individuals analysed) being shared by all seven populations while the other two haplotypes occurred only in Tabarka (h2: two individuals) and Kalatlandalos (h3: one individual) from the western Mediterranean basin. All populations showed very low values of haplotype and nucleotide diversities (Table 1). Because of the very low variation in 16S sequences, population genetics analyses were performed on COI sequences only.
Population structure
All pairwise F ST values within each Mediterranean region were very low (Table 2). However, all inter-regions pairwise comparisons yielded high values, with four of the 12 comparisons remaining significant after Bonferroni’s correction. The hierarchical AMOVA analysis revealed that 29% (P = 0.02) of the genetic variance was found among groups and 70.5% (P < 0.001) within populations (Table 3). However, the variance component explained by comparisons among populations within groups (0.5%) was low and not significant (P = 0.35).
There was no evidence of isolation by distance between the populations studied. The Mantel test detected no relationship between genetic and geographical distances (r = 0.019, P = 0.4). The statistical parsimony procedure yielded one network with unambiguous connections (Fig. 3). All COI haplotypes were closely connected and clustered according to their main area of occurrence. The high frequency and wide geographical distribution of haplotype 1 indicate that it is likely the ancestral haplotype.

Minimum-spanning network of the seven P. kerathurus COI haplotypes (1–7). The sizes of circles are proportional to Haplotype frequency. Perpendicular tick marks on the lines joining haplotypes represent the number of nucleotide substitutions
Discussion
Partial sequence for the 16S rRNA mitochondrial gene revealed very low variation between all P. kerathurus individuals analysed with a sequence divergence nearly equal to zero. This is consistent with previous observations on other penaeid species (Quan et al. 2001, 2004; Chu et al. 2003; De Francisco and Galetti 2005; Kumar et al. 2007) and contrasts with the relatively high diversity of this gene observed in many invertebrate marine species (Shearer et al. 2002 and references therein). 16S rRNA gene is known to occur in the conserved portion of the mtDNA (Meyer 1994) and has a low rate of evolution which makes it inaccurate for studying population differentiation but useful, as it has been shown in several cases, in characterizing cryptic decapod species (the spiny lobster Panulirus argus, Sarver et al. 1998; the pink shrimp Farfantepenaeus subtilis, Maggioni et al. 2001; the giant freshwater prawn Macrobrachium rosenbergii; De Bruyn et al. 2004 and the kuruma shrimp Penaeus japonicus, Tsoi et al. 2005). The homogeneity of the P. kerathurus populations seen in our study is probably related to the conserved nature of this gene in penaeids. In contrast to the situation with the 16S rRNA gene, the COI region sequences exhibited sufficient nucleotide diversity for population analysis. The sequence divergence values ranging from 0 to 1.37% were similar to those reported in various studies on other penaeid species investigated using COI gene sequencing (Baldwin et al. 1998; McMillen-Jackson and Bert 2003; Quan et al. 2001, 2004; De Francisco and Galetti 2005; Gusmão et al. 2006).
The main finding of this study was the population structuring across the distribution area studied. Two groups were identified on either side of the Siculo–Tunisian (S–T) strait, an eastern group and a western group. Populations within each group appeared to be panmictic and there was no evidence for an isolation-by-distance scheme of differentiation at the scale studied. Both groups showed group-specific haplotypes and restricted gene exchange (Fig. 2). For instance, the haplotype 2 was found in all eastern populations with moderate to high frequencies and absent in western populations. Furthermore, in the highly conservative 16S rRNA gene, we found two haplotypes, although with low frequencies, only in western populations. These genetic characteristics strongly affected the population pairwise F ST values and the AMOVA analyses. Thus, the F ST values between populations belonging to different groups were very high and significant for four comparisons. All intra-region comparisons were not significantly different from zero.
This genetic differentiation between eastern and western groups could be explained by hydrographic regimes in the region. In benthic species with long pelagic larval stages like P. kerathurus, water currents are assumed to play an important role in shaping the structure of genetic polymorphism. The water circulation along the North African coasts, characterized by a unidirectional east–south–east flow of currents (coming from the Atlantic) which leave the Tunisian coasts at the entry of the S–T strait, might therefore, constitute a physical barrier to larval dispersal. This may be plausible in our case, as gene flow could only be promoted by larvae drifting with currents, and also in the case of other species with similar life cycles in this region and in other regions where marine physical barriers were also suspected. For instance, genetic discontinuities have been noticed for bivalves (Quesada et al. 1995; Nikula and Vainola 2003) and seagrass (Arnaud-Haond et al. 2007) around the S–T strait; for sponge (Duran et al. 2004a, b), urchin (Duran et al. 2004c) and lobster (Triantafyllidis et al. 2005) around the strait of Gibraltar and for crab (Lavery et al. 1996) and prawn (Duda and Palumbi 1999) in the Indo-west Pacific. However, the same geographical regions do not necessarily restrict gene flow of other species with similar dispersal capabilities (Arculeo et al. 2003; Bargelloni et al. 2003, 2005; Zardoya et al. 2004; Patarnello et al. 2007 and references therein). We unfortunately cannot obtain a realistic estimate of gene flow by the use of indirect measure extrapolated from haplotype frequencies because our populations may not be at migration-drift equilibrium (Whitlock and McCauley 1999), as assumed by the model upon which the Wright’s (1931) equation [F ST ≈ 1/(2Nm + 1)] is based to estimate the absolute number of effective female migrants per generation (Nm). Even if we apply the Wright’s equation, the Nm-value between eastern and western groups (about 2) indicated that larval transportation on either side of the S–T strait may not be negligible. A common rule of thumb holds that Nm-values above 1 are sufficient to prevent the accumulation of fixed haplotype differences (Slatkin and Barton 1989). Moreover, substantial migration has been suggested for example in the European hake and the sea bass across the Almeria–Oran front (Naciri et al. 1999; Cimmaruta et al. 2005), which is a much more important oceanographic discontinuity than the S–T strait (Tintore et al. 1988; Pinardi and Masetti 2000). Therefore, water currents seem to be insufficient by themselves to explain the differentiation observed for certain species as genetic homogeneity is found in the same areas for other species with similar dispersal abilities. Overall, while present-day currents appear to be a good primary template for the establishment of genetic structure as the present data support, forces other than purely passive hydrological mechanisms may be also implied, such as homing behaviour and larval retention (Jones et al. 1999, 2005; Swearer et al. 1999) or selection against immigrants (Allegrucci et al. 1994, 1997; Lemaire et al. 2000). No data on larval retention are available for our species and little is known about its ecology. It is likely that further ecological and behavioural studies will provide further clues as to the reasons for the observed pattern of genetic structure.
The mtDNA variation in P. Kerathurus is consistent to some extent with an ancient split between eastern and western Mediterranean populations as suggested by the phylogenetic relationships between haplotypes. The minimum-spanning network demonstrated that the haplotypes, despite being closely connected and separated by few numbers of mutational steps, clustered to their main area of occurrence (Fig. 3). This suggests that populations on both sides of the S–T strait might be derived from one recent maternal ancestor. The overall mean nucleotide divergence between the two basins is 0.0025 ± 0.0017. Assuming a divergence rate for the COI gene of approximately 3% per million years (calibrated by the rise of the isthmus of Panama, Baldwin et al. 1998), the approximate divergence time is estimated to be about 82,000 years (±55,000), which suggests that the current populations of P. kerathurus might have diverged from a common lineage about 82,000 years ago. This timescale, which coincides with the later Pleistocene global glacial period, should be interpreted with caution since molecular clocks are known to be imprecise and sequence divergence rates may vary even within the same species (Zhang and Ryder 1995). During this period, climate fluctuations produced episodes of habitat fragmentation and reduced connectivity between eastern and western Mediterranean. The sea level recurrently dropped below the present-day level, reducing the width and depth of the S–T passage (Thiede 1978). Some genetic studies on other species showed that the S–T strait is an important genetic boundary between the eastern and western Mediterranean basins (Bahri-Sfar et al. 2000; Nikula and Vainola 2003; Arnaud-haond et al. 2007) and have highlighted the importance of such environmental fluctuations as an evolutionary force shaping population structure in this region. Concordance in the geographical positions of significant gene-tree partitions across multiple co-distributed species indicates that the same historical biogeographical factors likely influenced intraspecific patterns of genetic differentiation in these species.
We observed low mean values of haplotype and nucleotide diversities that are much lower than those reported for crustaceans (h > 0.8 and π > 0.01; see for examples Lavery et al. 1996; Stamatis et al. 2004; Inoue et al. 2007). Moreover, the haplotype relationship network is characterized by a nearly star genealogy centered on one geographically widespread haplotype. This phylogenetic pattern is commonly interpreted as a signature of a recent population expansion following a population bottleneck (Slatkin and Hudson 1991). Our results suggest that P. kerathurus populations may have undergone an historical bottleneck. A possible decline of population sizes can also explain the low genetic diversity but it is an unlikely cause because the population sizes are supposed to be large as it can be inferred from the catch sizes (the P. kerathurus landings in the studied area averaged 2,000 tons per year; Ben Abdallah et al. 2003). It is noteworthy that genetic diversity in populations from the western region was approximately 50% of that found in the eastern ones.
Ultimately, our study shows genetic differentiation between the eastern and western Mediterranean populations and suggests that divergent entities may meet in a contact zone between the gulf of Tunis and the gulf of Hammamet. The question is: how could such genetic disjunction be maintained? Is the presumed physical barrier (i.e. S–T strait) sufficient to limit gene flow? Is there any genetically determined barrier, resulting from population vicariance? Or do both barriers contribute in conjunction? If our estimate is realistic, the elapsed time since the divergence between eastern and western groups (around 82,000 years) seems insufficient for the establishment of reproductive isolating mechanisms. However, recent evidence from molecular genetics suggests diversification and speciation of some Indo-west Pacific marine taxa occurred as late as the Pleistocene (McMillan and Palumbi 1995; Palumbi 1996). We cannot conclude if this timescale is an underestimate or simply reflects very recent disjunction; the use of more rapidly evolving molecular markers such as microsatellites and/or mtDNA control region could help resolve this issue.
References
Allegrucci G, Fortunato C, Cataudella S, Sbordoni V (1994) Acclimation to fresh water of the sea bass: evidence of selective mortality and allozyme genotypes. In: Beaumont AR (ed) Genetics and evolution of marine organisms. Chapman and Hall, London, pp 486–502
Allegrucci G, Fortunato C, Sbordoni V (1997) Genetic structure and allozyme variation of seabass (Dicentrarchus labrax and D. punctatus) in the Mediterranean Sea. Mar Biol (Berl) 128:347–358. doi:10.1007/s002270050100
Arculeo M, Lo Brutto S, Sirna-Terranova M, Maggio T, Cannizzaro L, Parrinello N (2003) The stock genetic structure of two Sparidae species, Diplodus vulgaris and Lithognathus mormyrus, in the Mediterranean Sea. Fish Res 63:339–347. doi:10.1016/S0165-7836(03)00102-4
Arnaud-Haond S, Migliaccio M, Diaz-Almela E, Teixeira S, Van de Vliet MS, Alberto F, Procaccini G, Duarte CM, Serrão EA (2007) Vicariance patterns in the Mediterranean Sea: east–west cleavage and low dispersal in the endemic seagrass Posidonia oceanica. J Biogeogr 34:963–976. doi:10.1111/j.1365-2699.2006.01671.x
Avise JC (2000) Phylogeography: the history and formation of species. Harvard University Press, Cambridge
Bahri-Sfar L, Lemaire C, Ben Hassine OK, Bonhomme F (2000) Fragmentation of sea bass populations in the western and eastern Mediterranean as revealed by microsatellite polymorphism. Proc R Soc Lond B Biol Sci 267:929–935. doi:10.1098/rspb.2000.1092
Baldwin JD, Bass AL, Bowen BW, Wallis H, Clark JR (1998) Molecular phylogeny and biogeography of the marine shrimp Penaeus. Mol Phylogenet Evol 10:399–407. doi:10.1006/mpev.1998.0537
Bargelloni L, Alarcon JA, Alvarez MC, Penzo E, Magoulas A, Eis C, Patarnello T (2003) Discord in the family Sparidae (Teleostei): divergent phylogeographical patterns across the Atlantic-Mediterranean divide. J Evol Biol 16:1149–1158. doi:10.1046/j.1420-9101.2003.00620.x
Bargelloni L, Alarcon JA, Alvarez MC, Penzo E, Magoulas A, Palma J, Patarnello T (2005) The Atlantic-Mediterranean transition: discordant genetic patterns in two seabream species, Diplodus puntazzo (Cetti) and Diplodus sargus (L.). Mol Phylogenet Evol 36:523–535. doi:10.1016/j.ympev.2005.04.017
Belkhir K, Borsa P, Chikki L, Goudet J, Bonhomme F (2001) genetix v 4.03—Un logiciel pour l’analyse des données en génétique des populations. Université Montpellier II, France
Ben Abdallah O, Jarboui O, Missaoui H, Ben HadjHamida N (2003) Croissance relative, sex-ratio et exploration de la Crevette blanche Metapenaeus monoceros (Fabricius, 1798) du golfe de gabes (Tunisie). Bull Inst Natn Scien Tech Mer Salammbo 30:49–54
Benzi JAH, Williams ST (1997) Genetic structure of giant clam (Tridacna maxima) populations in the west Pacific is not consistent with dispersal by present-day ocean currents. Evol Int J Org Evol 51:768–783. doi:10.2307/2411153
Benzie JAH (2000) Population genetic structure in penaeid prawns. Aquacult Res 31:95–119. doi:10.1046/j.1365-2109.2000.00412.x
Borsa P, Naciri M, Bahri L, Chikhi L, Garcia de Leon FJ, Bonhomme F (1997) Zoogéographie infra-spécifique de la mer Méditerranée, analyse des données génétiques sur seize espèces atlanto-méditerranéennes (poissons et invertébrés). Vie Milieu 47:295–305
Chu KH, Li CP, Tam YK, Lavery S (2003) Application of mitochondrial control region in population genetic studies of the shrimp Penaeus. Mol Ecol Notes 3:120–122. doi:10.1046/j.1471-8286.2003.00376.x
Cimmaruta R, Bondanelli P, Nascetti G (2005) Genetic structure and environmental heterogeneity in the European hake (Merluccius merluccius). Mol Ecol 14:2577–2591. doi:10.1111/j.1365-294X.2005.02595.x
Cunningham CW, Collins T (1998) Beyond area relationships: extinction and recolonization in marine molecular biogeography. In: DeSalle R, Schierwater B (eds) Molecular approaches to ecology and evolution. Birkhauser-Verlag, Basel, pp 297–322
De Bruyn M, Wilson JA, Mather PB (2004) Huxley’s line demarcates extensive genetic divergence between eastern and western forms of the giant freshwater prawn, Macrobrachium rosenbergii. Mol Phylogenet Evol 30:251–257. doi:10.1016/S1055-7903(03)00176-3
De Francisco AK, Galetti PM (2005) Genetic distance between broodstocks of the marine shrimp Litopenaeus vannamei (Decapoda, Penaeidae) by mtDNA analyses. Genet Mol Biol 28:258–261
Duda TF, Palumbi SR (1999) Population structure of the black tiger prawn, Penaeus monodon, among western Indian and western Pacific populations. Mar Biol (Berl) 134:705–710. doi:10.1007/s002270050586
Duran S, Pascual M, Estoup A, Turon X (2004a) Strong population structure in the marine sponge Crambe crambe (Poecilosclerida) as revealed by microsatellite markers. Mol Ecol 13:511–522. doi:10.1046/j.1365-294X.2004.2080.x
Duran S, Pascual M, Turon X (2004b) Low levels of genetic variation in mtDNA sequences over the western Mediterranean and Atlantic range of the sponge Crambe crambe (Poecilosclerida). Mar Biol (Berl) 144:31–35. doi:10.1007/s00227-003-1178-5
Duran S, Palacin C, Becerro MA, Turon X, Giribet G (2004c) Genetic diversity and population structure of the commercially harvested sea urchin Paracentrotus lividus (Echinodermata, Echinoidea). Mol Ecol 13:3317–3328. doi:10.1111/j.1365-294X.2004.02338.x
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) arlequin version 3.0—an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
FAO (2006) Yearbook of fishery statistics: catches and landings. Food and Agriculture Organisation of the United Nations
Garcia S, Le Reste L (1981) Cycles vitaux, dynamique, exploitation et aménagement des stocks de crevettes penaeides côtières. FAO Doc Tech Pêches 203:210
Gusmão J, Lazoski C, Monteiro FA, Solé-Cava AM (2006) Cryptic species and population structuring of the Atlantic and Pacific seabob shrimp species, Xiphopenaeus kroyeri and Xiphopenaeus riveti. Mar Biol (Berl) 149:491–502. doi:10.1007/s00227-005-0232-x
Hellberg ME (1996) Dependence of gene flow on geographic distance in two solitary corals with different larval dispersal abilities. Evol Int J Org Evol 50:1167–1175. doi:10.2307/2410657
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc Lond 58:247–276
Inoue N, Watanabe H, Kojima S, Sekiguchi H (2007) Population structure of Japanese lobster Panulirus japonicas inferred by nucleotide sequence analysis of mitochondrial COI gene. Fish Sci 73:550–556. doi:10.1111/j.1444-2906.2007.01367.x
Johnson MS, Black R (1995) Neighbourhood size and the importance of barriers to gene flow in an intertidal snail. Heredity 75:142–154. doi:10.1038/hdy.1995.117
Jones GP, Millicich MJ, Lunow C (1999) Self-recruitment in a coral reef fish population. Nature 402:802–804. doi:10.1038/45538
Jones GP, Planes S, Thorrold SR (2005) Coral reef fish larvae settle close to home. Curr Biol 15:1314–1318. doi:10.1016/j.cub.2005.06.061
Krijgsman W, Hilgen FJ, Raffi I, Sierro FJ, Wilson DS (1999) Chronology, causes and progression of the Messinian salinity crisis. Nature 400:652–655. doi:10.1038/23231
Kumar N, Lakra WS, Majumdar KC, Goswami M, Ravinder K (2007) Genetic diversity in the Indian population of Penaeus monodon (Fabricius, 1798) as revealed by mtDNA sequence analysis. Aquacult Res 38:862–869. doi:10.1111/j.1365-2109.2007.01740.x
Lacson JM (1992) Minimal genetic variation among samples of six species of coral reef fishes collected at La Parguera, Puerto Rico, and Discovery Bay, Jamaica. Mar Biol (Berl) 112:327–331. doi:10.1007/BF00702479
Lavery S, Moritz C, Fielder DR (1996) Indo-Pacific population structure and evolutionary history of the coconut crab Birgus latro. Mol Ecol 5:557–570. doi:10.1111/j.1365-294X.1996.tb00347.x
Lavery S, Chan TY, Tam YK, Chuc KH (2004) Phylogenetic relationships and evolutionary history of the shrimp genus Penaeus s.l. derived from mitochondrial DNA. Mol Phylogenet Evol 31:39–49. doi:10.1016/j.ympev.2003.07.015
Lemaire C, Allegrucci G, Naciri M, Bahri-Sfar L, Kara H, Bonhomme F (2000) Do discrepancies between microsatellite and allozyme variation reveal differential selection between sea and lagoon in the sea bass (Dicentrarchus labrax)? Mol Ecol 9:457–467. doi:10.1046/j.1365-294x.2000.00884.x
Lemaire C, Versini JJ, Bonhomme F (2005) Maintenance of genetic differentiation across a transition zone in the sea: discordance between nuclear and cytoplasmic markers. J Evol Biol 18:70–80. doi:10.1111/j.1420-9101.2004.00828.x
Maggioni R, Rogers AD, Maclean N, D’Incao F (2001) Molecular phylogeny of western Atlantic Farfantepenaeus and Litopenaeus shrimp based on mitochondrial 16S partial sequences. Mol Phylogenet Evol 18:66–73. doi:10.1006/mpev.2000.0866
Magoulas A, Castilho R, Caetano S, Marcato S, Patarnello T (2006) Mitochondrial DNA reveals a mosaic pattern of phylogeographical structure in Atlantic and Mediterranean populations of anchovy (Engraulis encrasicolus). Mol Phylogenet Evol 3:734–746. doi:10.1016/j.ympev.2006.01.016
McMillan WO, Palumbi SR (1995) Concordant evolutionary patterns among Indo-west Pacific butterfly fishes. Proc R Soc Lond B Biol Sci 260:229–236. doi:10.1098/rspb.1995.0085
McMillen-jackson A, Bert TM (2003) Disparate patterns of population genetic structure and population history in two sympatric penaeid shrimp species (Farfantepenaeus aztecus and Litopenaeus setiferus) in the eastern United States. Mol Ecol 12:2895–2905. doi:10.1046/j.1365-294X.2003.01955.x
Meyer A (1994) DNA technology and phylogeny of fish. In: Beaumont AR (ed) Genetics and evolution of aquatic organisms. Chapman & Hall, London, pp 220–290
Naciri M, Lemaire C, Borsa P, Bonhomme F (1999) Genetic study of the Atlantic/Mediterranean transition in sea bass (Dicentrarchus labrax). J Hered 90:591–596. doi:10.1093/jhered/90.6.591
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nikula R, Vainola R (2003) Phylogeography of Cerastoderma glaucum (Bivalvia: Cardiidae) across Europe: a major break in the Eastern Mediterranean. Mar Biol (Berl) 143:339–350. doi:10.1007/s00227-003-1088-6
Ovenden JR, Brasher DJ, White RW (1992) Mitochondrial DNA analyses of the red rock lobster Jasus edwardsii supports an apparent absence of population subdivision throughout Australasia. Mar Biol (Berl) 112:319–326. doi:10.1007/BF00702478
Palumbi SR (1994) Genetic divergence, reproductive isolation, and marine speciation. Annu Rev Ecol Syst 25:547–572. doi:10.1146/annurev.es.25.110194.002555
Palumbi SR (1996) What can molecular genetics contribute to marine biogeography? An urchin’s tale. J Exp Mar Biol Ecol 203:75–92. doi:10.1016/0022-0981(96)02571-3
Palumbi SR, Grabowsky G, Duda T, Geyer L, Tachino N (1997) Speciation and population genetic structure in tropical Pacific sea urchins. Evol Int J Org Evol 51:1506–1517. doi:10.2307/2411203
Patarnello T, Volckaert FAMJ, Castilho R (2007) Pillars of Hercules: is the Atlantic–Mediterranean transition a phylogeographical break? Mol Ecol 16:4426–4444. doi:10.1111/j.1365-294X.2007.03477.x
Pinardi N, Masetti E (2000) Variability of the large scale general circulation of the Mediterranean Sea from observations and modelling: a review. Palaeogeogr Palaeoclimatol Palaeoecol 158:153–174. doi:10.1016/S0031-0182(00)00048-1
Planes S, Galzin R, Bonhomme F (1995) A genetic metapopulation model for reef fishes in oceanic islands: the case of the surgeonfish, Acanthurus triostetus. J Evol Biol 8:103–117
Quan J, Lu XM, Zhuang Z, Dai J, Deng J, Zhang Y-P (2001) Low genetic variation in Penaeus chinesis as relvealed by mitochondrial COI and 16s rRNA gene sequences. Biochem Genet 39:279–284. doi:10.1023/A:1010234816514
Quan J, Zhuang Z, Deng J, Dai J, Zhang Y-P (2004) Phylogenetic relationships of 12 penaeoidea shrimp species deduced from mitochondrial DNA sequences. Biochem Genet 42:331–345. doi:10.1023/B:BIGI.0000039808.12069.ed
Quesada H, Beynon CM, Skibinski DOF (1995) A mitochondrial DNA discontinuity in the mussel Mytilus galloprovincialis Lmk: Pleistocene vicariance biogeography and secondary intergradation. Mol Biol Evol 12:521–524
Rice WR (1989) Analyzing tables of statistical tests. Evol Int J Org Evol 43:223–225. doi:10.2307/2409177
Riginos C, Nachman MW (2001) Population subdivision in marine environments: the contributions of biogeography, geographical distance and discontinuous habitat to genetic differentiation in a blennioid fish, Axoclinus nigricaudus. Mol Ecol 10:1439–1453. doi:10.1046/j.1365-294X.2001.01294.x
Russo CAM, Solé-cava AM, Thorpe JP (1994) Population-structure and genetic-variation in two tropical sea-anemones (Cnidaria, Actinidae) with different reproductive strategies. Mar Biol (Berl) 119:267–276. doi:10.1007/BF00349566
Sarver SK, Silberman JD, Walsh PJ (1998) Mitochondrial DNA sequence evidence supporting the recognition of two subspecies or species of the Florida spiny lobster Panulirus argus. J Crustac Biol 18:177–186. doi:10.2307/1549532
Schmidt PS, Rand DM (1999) Intertidal microhabitat and selection at MPI: interlocus contrasts in the northern acorn barnacle, Semibalanus balanoides. Evol Int J Org Evol 53:135–146. doi:10.2307/2640926
Shearer TL, Van Oppen MJH, Romano SL, Wörheide G (2002) Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Mol Ecol 11:2475–2487. doi:10.1046/j.1365-294X.2002.01652.x
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P (1994) Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am 87:652–701
Slatkin M, Barton NH (1989) A comparison of three indirect methods for estimating average levels of gene flow. Evol Int J Org Evol 43:1349–1368. doi:10.2307/2409452
Slatkin M, Hudson RR (1991) Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129:555–562
Stamatis C, Triantafyllidis A, Moutou KA, Mamuris Z (2004) Mitochondrial DNA variation in northeast Atlantic and Mediterranean populations of Norway lobster, Nephrops norvegicus. Mol Ecol 13:1377–1390. doi:10.1111/j.1365-294X.2004.02165.x
Swearer SE, Caselle JE, Warner RR (1999) Larval retention and recruitment in an island population of a coral-reef fish. Nature 402:799–802. doi:10.1038/45533
Thiede J (1978) A glacial Mediterranean. Nature 276:680–683. doi:10.1038/276680a0
Thompson JD, Higgins DG, Gibson TJ (1994) clustal w: improving the sensitivity of progressive multiple sequence weighting, position-specific gap penalties and weight metric choices. Nucleic Acids Res 22:4673–4680. doi:10.1093/nar/22.22.4673
Tintore J, La Violette PE, Blade I, Cruzado A (1988) A study of an intense density front in the eastern Alboran Sea: the Almeria–Oran front. J Oceanogr 18:1384–1397. doi:10.1175/1520-0485(1988)018<1384:ASOAID>2.0.CO;2
Triantafyllidis A, Apostolidis AP, Katsares V, Kelly VE, Mercer J, Hughes M, Jørstad KE, Tsolou A, Hynes R, Triantaphyllidis C (2005) Mitochondrial DNA variation in the European lobster (Homarus gammarus) throughout the range. Mar Biol (Berl) 146:223–235. doi:10.1007/s00227-004-1435-2
Tsoi KH, Wang ZY, Chu KH (2005) Genetic divergence between two morphologically similar varieties of the kuruma shrimp Penaeus japonicus. Mar Biol (Berl) 147:367–379. doi:10.1007/s00227-005-1585-x
Uthicke S, Benzie JAH (2003) Gene flow and population history in high dispersal marine invertebrates: mitochondrial DNA analysis of Holothuria nobilis (Echinodermata: Holothuroidea) populations from Indo-Pacific. Mol Ecol 12:2635–2648. doi:10.1046/j.1365-294X.2003.01954.x
Waples RS (1987) A multispecies approach to the analysis of gene flow in marine shore fishes. Evol Int J Org Evol 41:385–400. doi:10.2307/2409146
Whitlock MC, McCauley DE (1999) Indirect measures of gene flow and migration: Fst # 1/(4Nm + 1). Heredity 82:117–125. doi:10.1038/sj.hdy.6884960
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Zardoya A, Castilho R, Grande C, Favre-Krey L, Caetano S, Marcato S, Krey G, Patarnello T (2004) Differential population structuring of two closely related fish species, the mackerel (Scomber scombrus) and the chub mackerel (Scomber japonicus), in the Mediterranean Sea. Mol Ecol 13:1785–1798. doi:10.1111/j.1365-294X.2004.02198.x
Zhang Y-P, Ryder OA (1995) Different rates of mitochondrial DNA sequences evolution in kirk’s dik–dik (Madoqua kirkii) populations. Mol Phylogenet Evol 4:291. doi:10.1006/mpev.1995.1026
Zitari-Chatti R, Chatti N, Elouaer A, Said K (2008) Genetic variation and population structure of the caramote prawn Penaeus kerathurus (Forskäl) from the eastern and western Mediterranean coasts in Tunisia. Aquacult Res 39:70–76
Acknowledgments
The authors thank Giorgio Bernardi for his interest in this work. They extend their thanks to Salvatore Cozzolino and Marina Prisco for helping in mtDNA sequencing of samples. We are grateful to the anonymous reviewers for helpful comments on the manuscript. This work was supported by a grant from the Tunisian Ministère de l’Enseignement Supérieur, de la Recherche Scientifique et de la Technologie.
Author information
Authors and Affiliations
Corresponding author
Additional information
R. Zitari-Chatti and N. Chatti are contributed equally to the work.
Rights and permissions
About this article
Cite this article
Zitari-Chatti, R., Chatti, N., Fulgione, D. et al. Mitochondrial DNA variation in the caramote prawn Penaeus (Melicertus) kerathurus across a transition zone in the Mediterranean Sea. Genetica 136, 439–447 (2009). https://doi.org/10.1007/s10709-008-9344-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-008-9344-9