
Abstract
Genetic testing of an Irish kindred identified an exonic nucleotide substitution c.1664T>C (p.Leu555Pro) in the MLH1 mismatch repair (MMR) gene. This previously unreported variant is classified as a “variant of uncertain significance” (VUS). Immunohistochemical (IHC) analysis and microsatellite instability (MSI) studies, genetic testing, a literature and online MMR mutation database review, in silico phenotype prediction tools, and an in vitro MMR activity assay were used to study the clinical significance of this variant. The MLH1 c.1664T>C (p.Leu555Pro) VUS co-segregated with three cases of classic Lynch syndrome-associated malignancies over two generations, with consistent loss of MLH1 and PMS2 protein expression on IHC, and evidence of the MSI-High mutator phenotype. The leucine at position 555 is well conserved across a number of species, and this novel variant has not been reported as a normal polymorphism in the general population. In silico and in vitro analyses suggest that this variant may have a deleterious effect on the MLH1 protein and abrogate MMR activity. Evidence from clinical, histological, immunohistochemical, and molecular genetic data suggests that MLH1 c.1664T>C (p.Leu555Pro) is likely to be the pathogenic cause of Lynch syndrome in this family.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lynch syndrome (MIM# 120435) is an inherited predisposition to a range of cancers, notably uterine cancer in females and colorectal cancer (CRC) in both sexes, caused by a deficiency in the DNA mismatch repair (MMR) pathway [1]. The MMR system consists of several nuclear proteins that act in concert to detect and repair replication errors (base–base mismatches and insertion–deletion loops). Cancer in Lynch syndrome develops because of the co-occurence of a pathogenic germline mutation in one of the highly-conserved MMR genes: mutS homolog 2 (MSH2) mutL homolog 1 (MLH1) mutS homolog 6 (MSH6) and postmeiotic segregation increased 2 (PMS2) and a subsequent somatic mutation involving the wild-type allele in a cell of the susceptible tissue. Large germline deletions encompassing the 3′ exons of tumour associated calcium signal transducer 1 (TACSTD1) also known as epithelial cell adhesion molecule (EPCAM) can additionally lead to epigenetic inactivation of the corresponding MSH2 allele within tissue actively expressing EPCAM resulting in Lynch syndrome [2, 3]. Transgenerational transmission of constitutional MLH1 promoter epimutation has also been described in rare Lynch syndrome cases [4]. Genetic counselling of at-risk individuals is optimally based on identification of the underlying deleterious germline mutation in an appropriate family member who has developed cancer. Pathogenic mutations are identified in approximately 60 % of microsatellite instability-high (MSI-H) cancer patients fulfilling clinical criteria for Lynch syndrome [5].
One outcome of genetic testing is the identification of a genetic variant of uncertain significance (VUS). These are often rare missense variants, which may represent neutral polymorphisms or may alter normal gene expression or function and be deleterious. Missense variants that disturb the DNA sequence and cause amino acid substitutions and silent exonic nucleotide substitutions are challenging to characterise. Some result in the substitution of one amino acid for another in the protein sequence, so-called non-synonymous substitutions, while others do not and are termed silent or synonymous codon substitutions. Approximately one-third of MMR gene VUSs are non-synonymous substitutions [6]. Pre-symptomatic genetic testing using a VUS to guide clinical care is not recommended. Determining which genetic variants are pathogenic and which are neutral is a major challenge in clinical genetics [7]. Various laboratory techniques have been applied towards characterising VUS. Establishing that the variant co-segregates with cancer incidence provides useful evidence of causation.
We identified a VUS in MLH1 c.1664T>C (p.Leu555Pro) in an Irish family. We present evidence to support our proposal that this MLH1 VUS should be designated as “likely pathogenic” (Class 4) according to the five class system proposed for assigning risk estimates to uncertain variants, comprising classifications: “pathogenic”, “likely pathogenic”, “uncertain”, “likely neutral”, depending on the available evidence [8]. We have integrated evidence from both direct (clinical) and indirect sources (data from in silico analyses and functional assays) [9].
Methods
Subjects
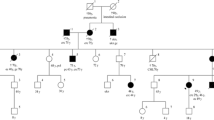
Data are presented on an Irish kindred (Fig. 1) which satisfies the Amsterdam II criteria [10], and within which an MLH1 VUS c.1664T>C (p.Leu555Pro) was identified through diagnostic genetic testing.
Members of this family received genetic counselling and provided peripheral blood samples for DNA extraction and genetic testing. All of the cases of malignancy described here were confirmed either by histopathology reports and/or death certificates. Informed consent was obtained for molecular studies on archival tissue.
Literature review
A review of the literature regarding this variant was undertaken and the following online sites were accessed: Human Gene Mutation database HGMD®, http://www.hgmd.cf.ac.uk/ac/index.php maintained by Cardiff University, International Society for Gastrointestinal Hereditary Tumors (InSiGHT) online MMR mutation database at www.insight-group.org/mutations which has been merged with the Mismatch Repair Gene Variants Database, www.med.mun.ca/MMRvariants, maintained by Memorial University in Newfoundland [11], and the Mismatch Repair Gene Unclassified Variants Database (www.mmruv.info), maintained by University Medical Center in Groningen [12] (accessed 16 and 17 March 2012).
In silico analysis was performed using the following web-based phenotype prediction tools: SIFT, Polyphen2, MAPP-MMR, Align GVGD, SNAP, Pmut and Mutation Taster.
Functional assay
A rapid next-generation cell-free assay was employed to quantify the repair efficiency of the variant MLH1 protein [13].
Clinical details
The index case became symptomatic and underwent an emergency sub-total colectomy for three synchronous colon cancers at the age of 23.
These tumours included a pT2 tumour arising in a tubulovillous adenoma at the mid portion of the specimen, a pT3 moderately differentiated caecal adenocarcinoma with 32 negative lymph nodes, and an early invasive pT1 carcinoma arising in a tubular adenoma in the caecum. Five pedunculated polyps in total were present within the caecum, and the other four polyps comprised tubulovillous adenomas without evidence of high grade dysplasia.
This presentation and the reported family history of cancer strongly suggested Lynch syndrome. A cancer genetic risk-assessment was initiated and a three-generation family history was recorded (Fig. 1).
The proband’s mother was diagnosed with ovarian and endometrial cancer at age 44. The endometrial pathology revealed atypical hyperplasia and a small fragment of early invasive endometrial adenocarcinoma. Sections of cystic ovary showed a borderline serous tumour. The other ovary showed a very high grade tumour with sarcomatous areas but mostly consisting of undifferentiated rhabdoid tumour. This ovarian pathology was felt to represent a very poorly differentiated adenocarcinoma of ovary or a poorly differentiated carcinosarcoma.
The proband’s maternal aunt underwent gynaecology screening because of her sister’s diagnosis and was diagnosed with a grade 1 endometrial adenocarcinoma, endometrioid type with associated focal complex endometrial hyperplasia at age 48. Two months post surgery, she became symptomatic and was diagnosed with a distal anterior rectal tumour which after abdominoperineal resection was classified as an infiltrating, moderately differentiated and extensive mucin producing adenocarcinoma. Eighteen lymph nodes showed reactive changes only. The proband’s maternal grandfather was diagnosed with gastric cancer in his mid 40s and died at age 51.
In vitro mismatch repair assays
Analysis of in vitro MMR activity was completed using a cell-free complementation assay as described by Drost et al. [13]. MLH1 c.1664T>C variant was generated by site-directed mutagenic PCR of wild type MLH1, cloned in pCITE4A, and directly expressed using the TnT Quick Coupled Transcription/Translation kit (Promega, Madison, WI, USA). Expressed VUS MLH1 protein was dimerised with in vitro produced wild-type PMS2, expressed from a pCITE4A-based expression construct. The heterodimeric protein-containing reticulocyte lysate was directly added to a plasmid substrate (pJHGT3′lnFAM) carrying a defined mismatching nucleotide (T/G) and an internal 6-FAM fluorescent label and nuclear extract of the human MLH1 and PMS2 deficient HCT-116 colon cancer cell line. Repair of the T/G mismatch to T/A by MMR restores a HinDIII restriction site, resulting in the generation of a 75-bp fluorescent diagnostic fragment. A 174-bp fragment represents an unrepaired substrate.
Results
Molecular investigations undertaken
Tumour blocks from the index case’s colon cancers were subjected to immunohistochemical (IHC) analysis for evidence of mismatch repair (MMR) protein expression. IHC staining demonstrated loss of expression of MLH1 and PMS2 MMR proteins.
Genetic testing
Following the IHC findings for the proband, sequencing of all nineteen exons and immediately-adjacent intronic regions of his MLH1 gene was completed in a Clinical Pathology Accredited (CPA) laboratory. This identified a thymine for cytosine (c.1664T>C) nucleotide substitution, that if translated, results in a proline for leucine amino acid substitution at position 555 of the MLH1 protein, in the ExoI interaction domain.
Multiplex ligation-dependent probe amplification (MLPA) of all nineteen exons of MLH1, all sixteen exons of MSH2 and exon 9 of EPCAM was also completed and showed no evidence of a gene deletion or duplication in the proband. There was no indication from in silico analysis that the identified sequence change affects splicing or created a novel high ranking splice/acceptor site, therefore RNA studies were not undertaken.
Co-segregation analysis
Tissue from the proband’s maternal aunt’s early onset rectal cancer demonstrated IHC loss of expression of the MMR proteins MLH1 and PMS2. Diagnostic sequencing of MLH1 in a different CPA accredited molecular laboratory also identified the c.1664T>C (p.Leu555Pro) VUS. MLH1 MLPA dosage analysis showed no evidence of a gene deletion or duplication.
The proband’s mother is an obligate heterozygote for the VUS, therefore IHC and MSI studies were undertaken on her archival tissue. Two paraffin blocks were submitted for analysis. Examination of hematoxylin and eosin (H&E) stained slides from the submitted blocks showed block 1 (labelled A1) represents a section of endometrium and myometrium in which the endometrium shows atypical hyperplasia with a focus of endometrial adenocarcinoma. The section submitted in block 2 (labelled A2) was described as a pelvic tumour and examination of the H&E stained slide showed a large mass of high grade tumour much of which is spindle cell with extensive necrosis. IHC on both tumours demonstrated complete loss of staining for the MMR proteins MLH1 and PMS2. Microsatellite instability (MSI) analysis showed the tumour in the uterus (A1) to be stable across all five markers analysed, while analysis of the pelvic mass tumour demonstrated MSI in three of five markers (MSI-H) analysed (mononucleotide markers BAT-26, BAT-25 and NR-27; see Fig. 2). BRAF mutation [14] status studies were completed on tumour tissue which was wildtype for the c.1799T>A (p.Val600Glu) activating mutation.
Review of literature and online databases regarding MLH1 c.1664T>C (p.Leu555Pro) unclassified variant
No record of this VUS was found on the following online databases; Human Gene Mutation database HGMD®, http://www.hgmd.cf.ac.uk/ac/index.php, or the International Society for Gastrointestinal Hereditary Tumors (InSiGHT) online MMR mutation database at www.insight-group.org/mutations, www.med.mun.ca/MMRvariants, or the Mismatch Repair Gene Unclassified Variants Database (www.mmruv.info).
In silico analysis of possible effects on protein function
The likely pathogenicity of VUS can be assessed by computational analysis. The following web-based in silico analyses exploit the relatively well understood protein multiple sequence alignments of the MLH1 gene across multiple species. Table 1 summarises the rationale and results of seven independent web-based algorithms that classify the MLH1 c.1664T>C (p.Leu555Pro) variant as being likely pathogenic.
In vitro mismatch repair assays
Analysis of in vitro MMR activity was completed using a cell-free complementation assay. The MLH1 c.1664T>C variant allele was reconstructed by PCR and variant protein was expressed in an in vitro transcription/translation kit directly from the PCR fragment. Variant MLH1 protein was dimerized with wild-type PMS2 protein produced by in vitro transcription/translation from a cloned wild type PMS2. The variant MLH1/PMS2 protein was added to an MLH1/PMS2 deficient extract together with a substrate with a defined mismatching nucleotide (T/G) and an internal 6-FAM fluorescent label. Proficiency of variant MLH1/PMS2 enables repair of the T/G mismatch to T/A by MMR, which results in the generation of a 75-bp fluorescent diagnostic fragment in addition to a 174-bp fluorescent fragment that represents unrepaired substrate. Conversely, loss of activity of variant MLH1/PMS will lead to absence of the diagnostic fragment, which is indicative of pathogenicity of the variant.
Variants G67R and I219V were produced in a similar fashion and included as pathogenic and polymorphic controls, respectively. In this assay, variant L555P repaired mismatches with an efficiency not significantly higher than the proven pathogenic MLH1 variant p.Gly67Arg (Fig. 3). The lack of MMR activity strongly supports the pathogenicity of variant p.Leu555Pro.
Discussion
We present a previously unreported MLH1 VUS in an Irish family and report data supporting its classification as pathogenic. Cumulative evidence from clinical, histological, immunohistochemical, bioinformatic and molecular data strongly suggest that the MLH1 c.1664T>A (p.Leu555Pro) variant is likely to be pathogenic resulting in Lynch syndrome.
We did not detect the c.1799T>A (p.Val600Glu) mutation in BRAF, which is frequently seen in sporadic MSI–High colorectal cancers with MLH1 hypermethylation, but not present in Lynch syndrome. The presence of BRAF c.1799T>A (p.Val600Glu) mutation in a tumour significantly reduces the likelihood of the presence of a germline mutation in either the MLH1 or MSH2 gene in Lynch syndrome associated colorectal cancers [14]. Wildtype BRAF identified in tumour tissue of an affected obligate carrier of the VUS in this kindred supports clinical presentation of Lynch syndrome.
This novel MMR variant has not been reported as a normal polymorphism in the general population. Missense mutations are more likely to be pathogenic when they occur in the interaction domains between MLH1 and PMS2, between the MutS and MutL heterodimers, or with ExoI [15]. The leucine at position 555 is located in the ExoI interaction domain and is well conserved across a number of species.
Conservation across species supports functional importance of a locus. Seven web based in silico phenotype prediction tools utilised in this study all predict MLH1 c.1664T>C (p.Leu555Pro) variant to be pathogenic (Table 1). These tools utilise algorithms based on publicly available protein alignments and, in some cases, basic structural information from the submitted amino acid sequence to determine pathogenicity. They are likely to be processing highly similar input information through somewhat similar algorithms, indicating that similar outputs should be treated more as recapitulation and re-assertion of the same body of evidence, as opposed to multiple individual data points. Nonetheless, there is robust consistency of the results from all seven prediction programs with the clinical and functional data.
An in vitro MMR assay revealed that the MLH1 c.1664T>A (p.Leu555Pro) variant has no higher MMR activity than a proven pathogenic MLH1 variant. The p.Leu555Pro substitution reduces the mismatch repair activity of MLH1 to a minimal level. As loss of MMR is strongly predictive for Lynch syndrome, the assay result provides strong evidence that the variant is pathogenic and causes Lynch syndrome. An integrative strategy such as the one presented here is required to adequately assess the clinical significance of a VUS, especially in the absence of segregation data from multiple families harbouring the same base pair substitution. Loss of heterozygosity (LOH) for the MLH1 wildtype allele in the tumour tissue may be investigated as a potential mechanism of tumourigenesis, however LOH data is excluded from multifactorial analysis until adequate reference data is established on the role of LOH in the development of cancer in carriers of MMR mutations [16, 17].
Due to the presence of several polyps in the index case at age 23, the possibility of either Attenuated Familial Adenomatous Polyposis (AFAP) or MUTYH–associated polyposis (MAP) as the potential disease pathway for this patient was also considered. Bi-allelic MUTYH mutations have been found in individuals with early onset CRC and few-to-no polyps [18], while hyperplastic polyps are common in MAP, albeit in a small study of 17 MAP patients where 8 of these individuals (47 %) displayed hyperplastic polyps and sessile serrated polyps [19]. Evidence that the MMR pathway is responsible for the pathogenesis of cancer in this family rather than the Base Excision Repair (BER) or APC pathway include the proband’s total polyp count which was less than 10 and the majority of polyps were not reported as either sessile serrated adenomas or hyperplastic, plus the consistent IHC loss of expression of the MMR proteins MLH1 and PMS2 in tumours from all three affected individuals. Therefore, screening for APC or biallelic MUTYH mutations was not undertaken. Although it remains possible that the patient could have bi-allelic MLH1 mutations/constitutional mismatch repair deficiency syndrome, there is no clinical evidence to support this, such as the presence of Cafe-au-lait spots, unusually early onset (diagnosed less than 20 years) and central nervous system or Lynch syndrome associated tumours.
Interestingly, the kindred presented shows evidence of anticipation, in which the age of onset of a disorder and/or the severity of the phenotype is increased in successive generations [20]. This phenomenon has been suggested in previous studies of Lynch syndrome [21–23] emphasising the importance of early-onset screening. However, convincing evidence for the anticipation effect in Lynch syndrome needs to be provided ideally in prospective studies, and the molecular mechanism underlying this phenomenon remains to be elucidated.
The IARC Working Group recommends that pre-symptomatic/predictive genetic testing be made available to family members of individuals with sequence variants of Classes 4 and 5 (likely to definitely pathogenic). Management recommendations for individuals harbouring a Class 4 variant are similar to those provided for Class 5 carriers. The evidence presented here supports the case for classification of this variant as Class 4 (Likely Pathogenic). The present strategy of multivariate analysis could be widely applied in clinical genetics to improve both the interpretation of VUS and the clinical management of patients harbouring these variants.
The evidence in support of pathogenicity is summarised as follows: (1) within this Amsterdam II positive Irish family the index case had clinically-defined Lynch syndrome with loss of expression of MLH1 and PMS2 proteins on IHC, (2) there was co-segregation of the variant with the most striking cancer phenotypes in two generations of the family, (3) including an obligate carrier, three individuals with classic Lynch syndrome phenotypes were shown to carry the VUS, with a young mean age of diagnosis of 38 years, (4) the variant was predicted to be pathogenic by several in silico assays, (5) functional analysis showed the complete loss of MMR activity in vitro. Finally, in support of our proposed classification, multifactorial likelihood analysis facilitated by the Variant Interpretation Committee of the International Society for Gastrointestinal Hereditary Tumors (InSiGHT) indicates that the posterior probability of pathogenicity of this variant is 0.97 which classifies c.1664T>C (p.Leu555Pro) as Class 4—Likely pathogenic [24].
The re-classification of the sequence change from VUS to “likely pathogenic” will allow pre-symptomatic testing to be offered to these family members and intensive lifelong cancer surveillance targeted to those in need.
MLH1 reference sequences
NM_000249.3 (mRNA sequence), NP_000240.1 (protein sequence).
References
Lynch HT, de la Chapelle A (2003) Hereditary colorectal cancer. N Engl J Med 348:919–932
Kovacs ME, Papp J, Szentirmay Z et al (2009) Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch Syndrome. Hum Mutat 30(2):197–203
Ligtenberg MJL, Kuiper RP, Chan TL et al (2009) Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet 41(1):114–117
Crépin M, Dieu M-C, Lejeune S et al (2012) Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptibility. Hum Mutat 33:180–188
Engel C, Forberg J, Holinski-Feder E et al (2006) Novel strategy for optimal sequential application of clinical criteria, immunohistochemistry and microsatellite analysis in the diagnosis of hereditary nonpolyposis colorectal cancer. Int J Cancer 118:115–122
Peltomaki P, Vasen HF (2004) Mutations associated with HNPCC predisposition—update of ICG-HNPCC/InSiGHT mutation database. Dis Markers 20:269–276
Rasmussen LJ, Heinen CD, Royer-Pokora B, Drost M, Tavtigian S, Hofstra RM, de Wind N (2012) Pathological assessment of mismatch repair gene variants in Lynch syndrome: past, present and future. Hum Mutat 33(12):1617–1625
Plon SE, Eccles DM, Easton D et al (2008) Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 29(11):1282–1291
Goldgar DE, Easton DF, Byrnes GB et al (2008) Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum Mutat 29:1282–1291
Vasen HF, Mecklin JP, Khan PM, Lynch HT (1991) The international collaborative group on hereditary non-polyposis colorectal cancer (ICG-HNPCC). Dis Colon Rectum 34:424–425
Woods MO, Williams P, Careen A et al (2007) A new variant database for mismatch repair genes associated with Lynch Syndrome. Hum Mutat 28(7):669–673
Ou J, Niessen RC, Vonk J et al (2008) A database to support the interpretation of human mismatch repair gene variants. Hum Mutat 29(11):1337–1341
Drost M, Zonneveld JBM, van Dijk L et al (2010) A cell-free assay for the functional analysis of variants of the mismatch repair protein MLH1. Hum Mutat 31:247–253
Domingo E, Laiho P, Ollikainen M et al (2004) BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet 41:664–668
Boland CR, Fishel R (2005) Lynch syndrome: form, function, proteins, and basketball. Gastroenterology 129(2):751–755
Spurdle AB (2013) Department of Genetics and Computational Biology, Queensland Institute of Medical Research, Brisbane, Australia (personal communication)
Hofstra RMW, Spurdle AB, Eccles D et al (2008) Tumor characteristics as an analytic tool for classifying genetic variants of uncertain clinical significance. Hum Mutat 29(11):1292–1303
Cleary SP, Cotterchio M, Jenkins MA et al (2009) Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology 136:1251–1260
Boparai KS, Dekker E, Van Eeden S et al (2008) Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology 135:2014–2018
Strachan T, Read AP (1999) Human molecular genetics. Wiley, New York
Westphalen AA, Russell AM, Buser M et al (2005) Evidence for genetic anticipation in hereditary non-polyposis colorectal cancer. Hum Genet 116:461–465
Stella A, Surdo NC, Lastella P et al (2007) Germline novel MSH2 deletions and a founder MSH2 deletion associated with anticipation effects in HNPCC. Clin Genet 71:130–139
Nilbert M, Timshel S, Bernstein I, Larsen K (2009) Role for genetic anticipation in Lynch Syndrome. J Clin Oncol 27:360–364
Spurdle AB, Thompson BA (2013) Department of Genetics and Computational Biology, Queensland Institute of Medical Research, Brisbane, Australia (personal communication)
Plotz G, Raedle J, Spina A et al (2008) Evaluation of the MLH1 I219V alteration in DNA mismatch repair activity and ulcerative colitis. Inflamm Bowel Dis 14(5):605–611
Author information
Authors and Affiliations
Corresponding author
Appendices
Appendix 1
See Fig. 1.

Amsterdam II positive Pedigree with MLH1 c.1664T>C (p.Leu555Pro) VUS
Appendix 2
See Fig. 2.

Microsatellite instability analysis of ovarian tissue DNA from obligate MLH1 c.1664 (p.Leu555Pro) carrier. (Color figure online)
Appendix 3
See Fig. 3.

MMR activity of MLH1 VUS as measured in the in vitro MMR assay. Results are shown as mean ± SEM of 4 fully independent experiments. Mock: Mock expression. I219V represents an innocuous polymorphism [25]. Asterisks Significantly higher than repair-deficient control G67R. (p < 0.001, student’s one-tailed t test). For “Mock” and “PMS2 only” reactions no repair was detected in all experiments
Rights and permissions
About this article
Cite this article
Farrell, M.P., Hughes, D.J., Drost, M. et al. Multivariate analysis of MLH1 c.1664T>C (p.Leu555Pro) mismatch repair gene variant demonstrates its pathogenicity. Familial Cancer 12, 741–747 (2013). https://doi.org/10.1007/s10689-013-9652-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-013-9652-9