
Abstract
Lynch syndrome is caused by germline mutations in any of the MisMatch Repair (MMR) genes. About 37% of MSH2 variants are missense variants causing single amino-acid substitutions. Whether missense variants affect the normal function of MMR proteins is crucial both to provide affected families a more accurate risk assessment and to offer predictive testing to family members. Here we report one family, fulfilling both Amsterdam I and II criteria and Bethesda guidelines, referred to our center for genetic counselling. The proband and some of her relatives have been investigated for microsatellite instability (MSI), immunohistochemical MMR protein staining, direct sequencing and Multiplex Ligation-dependent Probe Amplification (MLPA). Also Subcellular Localization Assay and Splice site predictions analyses were used. A germline missense variant of uncertain significance (exon 3, p.Val161Asp) was found in MSH2 gene in proband and in some relatives. The variant was associated with lack of expression of MSH2 protein (DMMR) and MSI-High status in tumour tissues. The localization assay of the MSH2 protein showed an abnormal subcellular localization pattern of the corresponding protein. Finally, splice-site prediction analysis ruled out a potential role of new splice sites as the cause behind the lack of expression of MSH2 protein and we suppose a potential correlation with other forms of post-transcriptional regulation (circular RNAs). The variant here reported shows a high correlation with phenotype and is located in an evolutionary conserved domain. The localization assay also suggest a potential pathogenic role, thus supporting further research on this matter.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lynch syndrome (LS), formerly known as hereditary non-polyposis colorectal cancer (HNPCC), is an autosomal dominant disorder predisposing to the development of colorectal cancers (about 80% lifetime risk) and of several other malignancies, including carcinomas of the endometrium, ovary, small bowel, stomach, ureter, biliary tract, pancreas, prostate, brain and some different types of skin cancers [1,2,3,4]. Early onset, proximal predominance in case of colorectal cancer, high number of synchronous and metachronous tumors, are usually associated with the syndrome.
LS is caused by a defect in any of the MisMatch Repair (MMR) genes, in particular MSH2, MLH1, MSH6 or PMS2, or by a deletion in the EPCAM gene, which leads to methylation of the adjacent MSH2 promoter [5]. These genes are responsible for repair of errors arising during DNA replication as a result of either incorrect base pairing or slippage of DNA polymerization on the template strand. Failure of DNA mismatch repair results in a mutator phenotype that leads to increased spontaneous somatic mutation rate, characterized by the change of the length of simple, repetitive nucleotide sequences that occur throughout the genome (microsatellites): this phenomenon is defined microsatellite instability (MSI) and represents a hallmark of this syndrome [6,7,8].
The early detection of individuals with LS is crucial for the identification of other mutation carriers in the same family who would benefit from risk-reduction strategies, such as cancer surveillance and prophylactic surgery.
Strategies for identifying LS-carriers include both the evaluation of personal and family cancer history, the application of molecular diagnostic testing of tumor tissue and, finally, the DNA mutational analysis.
Clinical criteria, represented by Amsterdam Criteria and Bethesda Guidelines, have been developed to identify individuals that, on the basis of their family history, might be candidates to receive genetic testing to assess presence of MMR genes germline pathogenic mutations [9,10,11,12,13].
Immunohistochemistry (IHC) for the expression of MLH1, MSH2, MSH6 and PMS2 proteins and MSI evaluation in tumour tissue play a key role as a screening method, providing informations for the presence or absence of proteins encoded by MMR genes, thus suggesting the specific gene defect (DMMR) [14, 15].
The presence of DMMR and MSI, together with family history, can be used to determine whether a patient could be a carrier of a MMR germline mutation, suggesting the need for further genetic tests [6, 14]: this is usually performed by using direct sequencing of entire MMR genes coding sequences and Multiplex ligation-dependent probe amplification (MLPA) analysis, evaluating the presence of large genomic rearrangements.
Mutation testing can provide three different “types” of results: pathogenic mutation, variant of uncertain significance (VUS), or informative negative finding, if no mutations are found [16].
Pathogenic variants may involve any area of each MMR gene and all known mutation types (frameshift, non-sense, splicing-site, missense variants and large gene rearrangements) could be responsible of defective MMR system [17], in absence of mutational “hot spots”.
Recently Plon et al. have classified MMR genes variants in to five classes: definitely pathogenic (class 5), likely pathogenic (class 4), of uncertain significance (class 3), likely not pathogenic or of little clinical significance (class 2) and not pathogenic or of no clinical significance (class 1) [16].
The majority of these variants are easily recognized as pathogenic as they result in the expression of truncated proteins. However, in LS families a substantial proportion of MLH1 and MSH2 variants are single nucleotide substitutions, either within coding sequences (missense, or silent mutations) or in intronic regions. Among them, about 28% of MLH1 and 37% of MSH2 variants are missense variants, which cause single amino-acid substitutions [18].
Whether these missense variants affect the normal function of MMR proteins, thus having a pathogenic role, is an essential information in genetic counselling, in order to provide affected families with a more accurate risk assessment but also to offer predictive (presymptomatic) genetic testing to family members.
Several criteria have been used to assess the pathogenicity of missense variants: co-segregation with the disease, low incidence in the general population, substitution of evolutionary preserved amino acids, non-conservative amino-acid changes and, in the case of suspected LS, correlation with MSI and IHC loss of MMR gene expression in tumour tissue [19, 20]. Here we report the case of a MSH2 missense variant, exon 3, c.482T>A, p.Val161Asp, classified in class 3 according to IARC classification [16] and found in a family fulfilling Amsterdam I criteria, Amsterdam II criteria and Bethesda guidelines, that on the basis of co-segregation with the disease, correlation with MSI, IHC loss of MMR protein expression in tumour tissue (DMMR) and localization assay, is strongly suggested having a pathogenic role.
Patients and methods
Family description
The proband (IV-9) was a 49-year-old female, whose personal and family history fulfilled both Amsterdam I and II criteria and Bethesda Guidelines for LS. At the age of 29 she was diagnosed with transverse colon adenocarcinoma and received subtotal colectomy with ileal-left colon anastomosis. At the age of 46 she developed a rectal adenocarcinoma (pT1N0M0) and, when she was 49, an endometrial adenocarcinoma (pT1N0M0). Then she was referred to our Center.
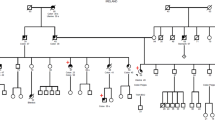
Detailed patient family history was collected: pedigree was drawn until fourth generation. All relatives’ data collected included sex, tumour site, age at diagnosis, together with current age or age of death (Fig. 1).

Famiy pedigree—the arrow indicates the proband. The type of tumour, as well as the age of onset, are describes. cr Colorectal cancer, gc gastric cancer, bc breast cancer, ec endometrial cancer, po polyps, pc prostate cancer, CML chronic myeloid leukemia, rc rectal cancer, uc urothelial cancer, btc biliary tract cancer
Patient’s family showed a typical LS pedigree. Proband’s mother (III-9) developed colon cancer when she was 58 and rectal cancer at the age of 71, while proband’s brother (IV-10) died at the age of 36 due to a biliary tract adenocarcinoma.
Among nine maternal relatives, one uncle had prostate cancer at 63 and colon cancer at 73 (III-5), one aunt was diagnosed with colon and breast cancer at the age of 50 (III-1), another aunt had endometrial cancer at 40 and gastric cancer at 70 (III-3), another one developed endometrial cancer when she was 60 years (III-2), while her son had a urothelial cancer at the age of 45 (IV-2) (Fig. 1). Risk assessment was also implemented by the result of the prediction of mismatch repair gene mutations in MLH1, MSH2 and MSH6 (PREMM 1,2,6) model, which estimated an individual probability of carryng a MMR genes mutation of 77.9%.
Proband’s tumour specimens were available at the Department of Pathology of our Institution and were examined by one pathologist.
Microsatellite instability analysis (MSI)
MSI was performed for the proband (IV-9) and relative (III-9).
Genomic DNA was isolated from tumour samples and corresponding normal tissues as previously described [21]. Tumours were examined for MSI using the 5-marker panel (two mononucleotide repeats––BAT25 and BAT26 and three dinucleotide repeats––D2S123, D5S346 and D17S250) recommended by the National Cancer Institute Workshop on MSI for Cancer Detection and Familial Predisposition. Tumours were classified as highly unstable (MSI-H) if at least 40% of the markers showed instability [6].
Oligonucleotide primers were fluorescently labelled (5′-VIC, 5-’NED, 5′-6-FAM, 5′-6-FAM, 5′-PET), and PCR products were evaluated with an using 3500DX Genetic Analyser based on automated capillary electrophoresis and automated sizing of the alleles by GeneScan 3.7 (Life Technologies, Foster City, CA). CAT25 microsatellite was also studied in all subjects [22].
Expression of MLH1, MSH2 and MSH6 proteins in tumour tissues
Tumour slides were available for the proband’s (IV-9) endometrial cancer (Fig. 2) and for relative III-9 rectal cancer.
Expression of the MLH1, MSH2 and MSH6 proteins was detected by IHC on 2-µm sections of formalin-fixed, paraffine embedded tissues, following antigen retrieval as previously described [23]. Primary antibodies used for IHC were: anti-MLH1 protein: clone G168–728, PharMingen, San Diego, CA, 1:50 dilution; anti-MSH2: clone FE11, Oncogene Research Products, Cambridge, MA, 1:100 dilution; anti-MSH6: clone H-141: sc-10798; Santa Cruz Biotechnology, Inc. Santa Cruz, CA 1:250 dilution.
Mutation analysis of the MLH1, MSH2 and MSH6 genes
Mutation analysis was performed on genomic DNA, isolated from peripheral blood lymphocytes, according to standard procedures. The 19 MLH1 exons, 16 MSH2 exons and 10 MSH6 exons, including flanking intronic regions, were individually amplified and directly sequenced using 3500DX Genetic Analyzer and the Big Dye terminator cycle sequencing Ready Reaction-Kit v. 3.1 (Life Technologies) [21]. The identification of DNA sequence variants has been confirmed by repeat PCR and sequence analysis of both DNA strands. Identified mutations were confirmed on a second sample PCR product. Primer sequences are available from the corresponding Author upon request. Our results were compared with MLH1, MSH2 and MSH6 normal sequence (respectively available at HNPCC database, http://www.insight-group.org).
Multiplex ligation-dependent probe amplification (MLPA) analysis
Patients studied for MLH1, MSH2, MSH6 sequence were also studied with Multiplex Ligation dependent Probe Amplification (MLPA) analysis.
MLPA was performed with 75 ng of an healthy control and patient DNAs, using the MRC-Holland (Amsterdam, Holland) MLH1, MSH2 and MSH6 SALSA MLPA Kits, according to the supplier’s protocol, run on an automatic ABI310 DNA analyzer, and evaluated with GeneScan sofware (Applied Biosystems). The electropherograms showed specific peaks corresponding to each exon of MSH2, MLH1 and MSH6 as well as additional peaks corresponding to reference sequences mapping on different chromosomes. A 40–55% decrease of the area of an MSH2 or MLH1 or MSH6 exon peak compared to the wild-type control samples was considered as indicative of an heterozygous deletion.
In silico prediction models
In silico tools were applied to estimate “a priori” probability of pathogenicity for the observed variant. The multivariate analysis of protein polymorphisms that focuses on the MRR genes (MAPP-MMR) carried out a score of 27.310. This result undoubtedly represents a large deviation with high probability of protein functional impairment, confirmed also by the PolyPhen-2 (Polymorphism Phenotyping v2) output of 0.99 [24, 25].
Protein subcellular localization assay
The p.Val161Asp MSH2 non-truncating variant was analyzed, in a homologous expression system, for possible abnormalities in nucleo-cytoplasmic shuttling through the assessment of the sub-cellular localization of the corresponding mutated protein in order to pinpoint the nuclear import impairment of the protein as a possible causative mechanism of MMR deficiency and LS susceptibility. The entire MSH2 (GenBank NM_000251) encoding region was RT-PCR amplified from the MMR-competent 293 human cell line and directionally cloned into the pEGFP-N1 eucariotic expression vector (BD Biosciences, Palo Alto, CA) between the XhoI and BamHI restriction sites and in frame with the amino terminus of the enhanced green fluorescent protein (EGFP) encoding gene. The resulting construct expressing MSH2-EGFP fluorescent fusion protein is reported here as pEGFP-MSH2. The mutant MSH2 derivative pEGFP-MSH2 (p.Val161Asp) was generated to the pEGFP-MSH2 plasmid between the XhoI and HindIII sites by PCR based site-directed mutagenesis method [26]. The mutation p.Pro622Leu was included in the study as putative impairing sub-cellular localization control. The introduction of the specific mutations and the absence of undesired second site sequence alterations were verified by direct DNA sequencing (Abi Prism 310 Genetic Analyzer—Applied Biosystem, Foster City, CA) prior to further use. For the expression of WT or mutant MSH2-EGFP fluorescent fusion proteins in a suitable homologue system, 0.5 × 106 MMR-proficient 293 cells were transiently transfected with 2 μg of pEGFP-MSH2 (WT or mutant) recombinant expression vectors. For fluorescent MSH2-EGFP fusion proteins detection, the 293 cells were fixed, 24 h after transfection, and stained with a 1 μg/ml solution of the DNA intercalating specific dye 4′,6-diamidino-2-phenylIndole (DAPI) (Sigma Aldrich, St Louis, MO). Sub-cellular localization of MSH2-EGFP recombinant proteins was analyzed by direct fluorescence using an Imager A1 microscope with a 63× objective (Zeiss, Thornwood, NY). Representative images were captured with Axio Cam MRC5 and processed with Axio Vision software (Zeiss, Thornwood, NY). At least 100 cells, from each of the three individual transfections performed, were analyzed from randomly sampled microscope fields of view.
Splice site predictions
SplicAid2 tools [27] was used to predict if mutated MSH2 RNA compared to the original one had different pattern of bound splicing proteins: the binding of a silencer factor in an exon could cause alternative splicing in an otherwise constitutively expressed exon or even cause exon skipping. On the other hand, gain of an enhancer sequence in an exon is usually irrelevant since these factors are already quite abundant in exons.
To predict if the mutation can generate or remove 5′ and 3′ splice sites, NNSPLICE tool [28] was used. A brief guide to interpret the consequences of the splicing site alteration events was reported in the paper cited here [29].
Since the protein abundance depends also by the efficiency of nucleocytoplasmic export of the respective transcript, we used ExportAid tool [30] to detect if the mutation alters, creates or destroys mRNA export elements in MSH2 gene: nucleocytoplasmic export of a transcript can be facilitated, hampered or even prevented by specific elements as, for example, the human eIF4E Sensitive Element (eIF4E-SE), the cytoplasmic accumulation region (CAR) or the constitutive transport element (CTE).
Results
Microsatellite instability analysis
Proband (IV-9) and III-9 relative showed high microsatellite instability (MSI-H): two out the five loci studied (Bethesda panel) resulted altered (40% MSI), BAT25 and BAT26.
Also The CAT25 microsatellite analysis showed instability in tested relative with MSI-H (Table 1).
Expression of MLH1, MSH2 and MSH6 proteins in tumour tissues
Study of the proteins expression was conducted in tumour specimens of the proband (IV-9) and in tumor specimens of the III-9 relative: normal staining for MLH1 and the lack of expression of MSH2 and MSH6 was observed (Fig. 2; Table 1).

Immunohistochemical staining of MMR proteins evaluated on endometrial carcinoma of the proband (IV-9). a Normal nuclear expression of MLH1 protein. b Lack of expression of MSH2 protein. c Lack of expression of MSH6 protein
Mutation analysis and MLPA of the MLH1, MSH2 and MSH6 genes
The sequence analysis of the entire coding region of MLH1 and MSH6 did not show any pathogenic variant.
A germline missense variant p.Val161Asp in exon 3 of MSH2 was found in proband (IV-9) and in relatives III-9, III-10, IV-2, IV-11, V-1, V-2.
The p.Val161Asp causes the substitution of a Thymine with an Adenine at nucleotide 482, leading a substitution of a Valine, which is a neutral non-polar amino acid, with Aspartic Acid, which is an acidic polar amino acid.
This variant is reported in ICG-HNPCC/InSiGHT mutation database (http://www.insightgroup.org/)and classified as class 3 (uncertain significance) according to IARC classification [16].
MLPA analysis did not show any evidence of exonic large deletions in all the subject analyzed (Table 1).
In silico prediction models
Even though InSiGHT database classifies this mutation as class 3 (uncertain), the “a priori” probability of this mutation being pathogenic when performing calculations by using Polyphen 2.0 [24] was 99% and MAPP-MMR score was 27.310 [25], thus suggesting a very high likelihood of the variant being pathogenic.
Furthermore, PREMM model reported the likelihood of having pathogenic mismatch repair mutations in this family, based on pedigree informations, of 77.9%.
The variant here reported is localized in a conserved domain (http://blocks.fhcrc.org/sift/SIFT.html; http://tux.embl-heidelberg.de/ramensky/polyphen.cgi).
Sub-cellular localization assay
The EGFP-MSH2 (WT) fusion protein was correctly imported through the nuclear pore complex (NPC) and detected almost completely in the nuclear district, whereas the EGFP-MSH2 (p.Pro622Leu) one, known as a nuclear import impairing variant [31], co-expressed with endogenous MSH6, was mainly located in the cytoplasm and showed a diffuse nuclear-cytoplasmic distribution pattern.
The EGFP-MSH2 (p.Val161Asp) variant was evenly spread out among nuclear and cytoplasmic compartments and associated with an abnormal subcellular localization pattern of the corresponding fusion protein (Fig. 3).

Localization analysis of recombinant wild type and mutated EGFP-MSH2 fusion proteins transiently expressed in 293 MMR proficient cells. Nuclei were stained with 4′,6-diamino-2-phenylindole (DAPI) and detected by direct fluorescence analysis 24 h after transfection. a Nuclear expression of EGFP-MSH2 (WT) (normal localization control). b Mainly cytoplasmic expression of EGFP-MSH2 (P622L) (abnormal localization control). c Mainly cytoplasmic expression of EGFP-MSH2 (V161D) mutation similar to abnormal localization control (original magnification ×630)
Splice site prediction
According to SpliceAid2 tool, the mutation creates a binding site for SRp30c and SC35 splicing proteins. Generally these factors contribute to exon definition but since this area is already an exon, their binding should not alter the splicing process. New splice sites are not created according to NNSPLICE tool. According to ExportAid tool, the mutation does not create motifs regulating nuclear export of the MSH2 transcript. The variation lies in a locus that gives rise to three circular RNAs (circRNAs) according to circBase repository (http://www.circbase.org/): hsa_circ_0006402, hsa_circ_0054469 and hsa_circ_0054470.
Discussion
Lynch syndrome is a hereditary form of colorectal cancer associated with germline deleterious defects in post-replicative DNA MMR genes, mainly MLH1, MSH2 and MSH6 [1, 4].
LS genetic testing deals with the identification of healthy carriers who can be advised about the risk of colorectal cancer and to act risk-reducing strategies, such as cancer surveillance and prophylactic surgery.
While the deleterious effect of truncating mutations and large rearrangement is well established (and easily conceivable on the basis of the genetic defect), the clinical significance of most non-truncating variants, such as missense, can be more difficult.
A significant fraction (20–25%) of LS related MSH2 germline variants deals with single aminoacid changes often associated with atypical clinical phenotypes [5, 17, 32]. When linkage analysis is not feasible and the biochemical significance of the alteration is uncertain, the nature of the mutation should be functionally characterized before confidently assigning it a pathogenic role.
Immunohistochemical analysis of MMR protein status in the tumor and MSI status can provide useful clues to identify which MMR gene is involved in tumour pathogenesis: indeed, the lack of expression of one of the proteins or the demonstration of MSI-High status might also be relevant in suggesting a correlation with a pathogenic mutation. Moreover, immunostaining may help to solve the role of MSH2 germline variants of uncertain significance because somatic inactivation of MSH2 is a rare event in sporadic microsatellite unstable tumours [33].
Here we report a MSH2 germline missense variant caused by substitution of a Valine with a Aspartate, c.482T>A, p.Val161Asp, in exon 3.
The analyzed family fulfills Amsterdam I and II criteria and Bethesda Guidelines. All affected family members, who underwent genetic tests, carried the same missense MSH2 variant in exon 3.
The variant here reported was not found in 90 healthy individuals, while all family members that developed tumours were mutation carriers, showing a high correlation with phenotype.
Even though InSiGHT database classifies this mutation as class 3 (uncertain), the “a priori” probability of this mutation being pathogenic when performing calculations by using Polyphen 2.0 was 99% and MAPP-MMR score was 27.310, thus suggesting a very high likelihood of the variant being pathogenic [24, 25].
Furthermore, PREMM model reported the likelihood of having pathogenic mismatch repair mutations in this family, based on pedigree informations, of 77.9%.
The T to A transition leads to an amino acid change in which a Valine, which is a non-polar amino acid, is substituted by an Aspartic Acid residue. The change is located in an evolutionary conserved domain and may indicate the functional relevance of this amino acid for the structure or function of the protein.
In another previous paper we described the possibility to assess the pathogenic role of MSH2 missense mutations of unknown significance using a subcellular localization assay. The latter, performed in a human homologous system, reliably classifies as pathogenic two MSH2 non-truncating alterations (p.Gly162Arg and p.Arg359Ser), providing an useful tool to assess MSH2 variants pathogenicity [7], similarly to the results of our present work.
Indeed, in the MSH2 protein localization assay, the EGFP-MSH2 (p.Val161Asp) variant, was associated with a diffuse nuclear-cytoplasmic distribution pattern of the corresponding fusion protein similar to the abnormal subcellular localization pattern of the control (Fig. 3).
Furthermore, both proband’s (IV-9) and her mother tumours’ specimens (III-9) show MSI-H status and lack of expression of MSH2 protein. Unfortunately it was not possible to perform microsatellite and IHC analysis in the other affected members because the tumour tissue was unavailable.
When we evaluated whether this change in MSH2 protein expression might be explained by different splice site alteration, the tools that we used seemed to rule out this possibility.
Our hypothesis is that another mechanism that might cause this impaired expression of the protein might be related to another form of post-transcriptional regulation, as in the form of circulating microRNAs. In particular, since the mutation lies in a locus that originates three different circular RNAs (circRNAs) we speculate that the alteration of circRNA sequence could somehow have pathological effects and our data suggest a new direction to investigate.
Circular RNAs (circRNAs) are endogenous noncoding RNAs that are formed upon splicing events through exon or intron circularization [34, 35]. Their covalently closed loop structure provides resistance against RNA exonuclease and good stability [36,37,38]. CircRNAs have multiple roles, initially it was discovered that they could act as microRNA sponges and therefore can regulate gene expression [39, 40]. Subsequently it emerged that they can bind proteins, for example, circ-Foxo3 circRNA binds Foxo3 and p53 resulting in an overall Foxo3 increase and p53 decrease [41]; they can sequester proteins, for example, intronic lariats in the cytoplasm sequester TDP-43 protein [42]; they can modulate gene expression, for example, EIciEIF3J and EIciPAIP2 circRNAs enhanced transcription levels of the corresponding EIF3J or PAIP2, their parental genes in cis [43]. CircRNAs are interesting also because they have a tissue-specific expression and their alteration is associated to human neurodegenerative [44] and tumour diseases [45, 46].
The reclassification of this variant as deleterious has significant implications in the management of this family and we suggest that could be relevant also for other people who are offered genetic testing.
Our findings also suggest the importance of combining genetic and clinical observations with data from localization assays to assess whether a missense variant is likely to be responsible for cancer development in LS families: clarifying the pathogenic role of missense mutations may increase the effectiveness of genetic testing programs and facilitate the planning of appropriate preventive strategies of high risk mutation carriers from family segregating such variants.
References
de la Chapelle A (2005) The incidence of Lynch syndrome. Fam Cancer 4(3):233–237. doi:10.1007/s10689-004-5811-3
Lynch HT, de la Chapelle A (2003) Hereditary colorectal cancer. N Engl J Med 348:919–932. doi:10.1056/NEJMra012242
Haraldsdottir S, Hampel H, Wei L, Wu C, Frankel W, Bekaii-Saab T, de la Chapelle A, Goldberg RM (2014) Prostate cancer incidence in males with Lynch syndrome. Genet Med 16:553–5577. doi:10.1038/gim.2013.193
Peltomäki P (2005) Lynch syndrome genes. Fam Cancer 4:227–232. doi:10.1007/s10689-004-7993-0
Vasen HF, Blanco I, Aktan-Collan K, Gopie JP, Alonso A, Aretz S, Bernstein I, Bertario L, Burn J, Capella G, Colas C, Engel C, Frayling IM, Genuardi M, Heinimann K, Hes FJ, Hodgson SV, Karagiannis JA, Lalloo F, Lindblom A, Mecklin JP, Møller P, Myrhoj T, Nagengast FM, Parc Y, Ponz de Leon M, Renkonen-Sinisalo L, Sampson JR, Stormorken A, Sijmons RH, Tejpar S, Thomas HJ, Rahner N, Wijnen JT, Järvinen HJ, Möslein G, Mallorca Group (2013) Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut 62:812–823. doi:10.1136/gutjnl-2012-304356
Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S (1998) A National Cancer Institute Workshop on microsatellite instability for cancer detection and familial predisposition: development on international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 58:5248–5257
Belvederesi L, Bianchi F, Galizia E, Loretelli C, Bracci R, Catalani R, Amati M, Cellerino R (2008) MSH2 missense mutations and HNPCC syndrome: pathogenicity assessment in a human expression system. Hum Mutat 29(11):E296–E309. doi:10.1002/humu.20875
Maccaroni E, Bracci R, Giampieri R, Bianchi F, Belvederesi L, Brugiati C, Pagliaretta S, Del Prete M, Scartozzi M, Cascinu S (2015) Prognostic impact of mismatch repair genes germline defects in colorectal cancer patients: are all mutations equal? Oncotarget 6(36):33848–38737. doi:10.18632/oncotarget.5395
Vasen HF, Mecklin JP, Khan PM, Lynch HT (1991) The International Collaborative Group on hereditary non-polyposis colorectal cancer (ICG-HNPCC). Dis Colon Rectum 34(5):424–425
Vasen HF, Watson P, Mecklin JP, Lynch HT (1999) New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 116(6):1453–1456
Syngal S, Fox EA, Eng C, Kolodner RD, Garber JE (2000) Sensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J Med Genet 37(9):641–645
Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, Lynch H, Perucho M, Smyrk T, Sobin L, Srivastava S (1997) A National Cancer Institute Workshop on hereditary nonpolyposis colorectal Cancer syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst 89(23):1758–1762
Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HF, Hawk ET, Barrett JC, Freedman AN, Srivastava S (2004) Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 96(4):261–268
Strate LL, Syngal S (2005) Hereditary colorectal cancer syndromes. Cancer Causes Control 16:201–213. doi:10.1007/s10552-004-3488-4
Bianchi F, Galizia E, Porfiri E, Belvederesi L, Catalani R, Loretelli C, Bracci R, Bearzi I, Turchi C, Viel A, Cellerino R (2007) A missense germline mutation in exon 7 of the MSH2 gene in a HNPCC family from center-Italy. Fam Cancer 6(1):97–102. doi:10.1007/s10689-006-9110-z
Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Hogervorst FB, Hoogerbrugge N, Spurdle AB, Tavtigian SV; IARC Unclassified Genetic Variants Working Group (2008) Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 29:1282–1291. doi:10.1002/humu.20880.
Peltomäki P, Vasen H (2004) Mutations associated with HNPCC predisposition—Update of ICG-HNPCC/INSiGHT mutation database. Dis Markers 20(4–5):269–276
Plazzer JP, Sijmons RH, Woods MO, Peltomäki P, Thompson B, Den Dunnen JT, Macrae F (2013) The InSiGHT database: utilizing 100 years of insights into Lynch syndrome. Fam Cancer 12(2):175–180. doi:10.1007/s10689-013-9616-0
Cravo M, Afonso AJ, Lage P, Albuquerque C, Maia L, Lacerda C, Fidalgo P, Chaves P, Cruz C, Nobre-Leitão C (2002) Pathogenicity of missense and splice site mutations in hMSH2 and hMLH1 mismatch repair genes: implications for genetic testing. Gut 50:405–412
Genuardi M, Carrara S, Anti M, Ponz de Leon M, Viel A (1999) Assessment of pathogenicity criteria for constitutional missense mutations of the hereditary nonpolyposis colorectal cancer genes MLH1 and MSH2. Eur J Hum Genet 7:778–782. doi:10.1038/sj.ejhg.5200363
Scartozzi M, Bianchi F, Rosati S, Galizia E, Antolini A, Loretelli C, Piga A, Bearzi I, Cellerino R, Porfiri E (2002) Mutations of hMLH1 and hMSH2 in patients with suspected hereditary nonpolyposis colorectal cancer: correlation with microsatellite instability and abnormalities of mismatch repair protein expression. J Clin Oncol 20:1203–1208. doi:10.1200/JCO.2002.20.5.1203
Bianchi F, Galizia E, Catalani R, Belvederesi L, Ferretti C, Corradini F, Cellerino R (2009) CAT25 is a mononucleotide marker to identify HNPCC patients. J Mol Diagn 11(3):248–252. doi:10.2353/jmoldx.2009.080155
Marcus VA, Madlensky L, Gryfe R, Kim H, So K, Millar A, Temple LK, Hsieh E, Hiruki T, Narod S, Bapat BV, Gallinger S, Redston M (1999) Immunohistochemistry for hMLH1 and hMSH2: a practical test for DNA mismatch repair-deficient tumors. Am J Surg Pathol 23:1248–1255
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249. doi:10.1038/nmeth0410-248
Drost M, Lützen A, van Hees S, Ferreira D, Calléja F, Zonneveld JB, Nielsen FC, Rasmussen LJ, de Wind N (2013) Genetic screens to identify pathogenic gene variants in the common cancer predisposition Lynch syndrome. Proc Natl Acad Sci USA 110(23):9403–9408. doi:10.1073/pnas.1220537110
Nyström-Lahti M, Perrera C, Räschle M, Panyushkina-Seiler E, Marra G, Curci A, Quaresima B, Costanzo F, D’Urso M, Venuta S, Jiricny J (2002) Functional analysis of MLH1 mutations linked to hereditary nonpolyposis colon cancer. Genes Chromosom Cancer 33(2):160–167
Piva F, Giulietti M, Burini AB, Principato G (2012) SpliceAid 2: a database of human splicing factors expression data and RNA target motifs. Hum Mutat 33:81–85. doi:10.1002/humu.21609
Reese MG, Eeckman FH, Kulp D, Haussler D (1997) Improved splice site detection in Genie. J Comput Biol 4:311–323
Piva F, Giulietti M, Nardi B, Bellantuono C, Principato G (2010) An improved in silico selection of phenotype affecting polymorphisms in SLC6A4, HTR1A and HTR2A genes. Hum Psychopharmacol 25:153–161. doi:10.1002/hup.1100
Giulietti M, Milantoni SA, Armeni T, Principato G, Piva F (2015) ExportAid: database of RNA elements regulating nuclear RNA export in mammals. Bioinformatics 31:246–251. doi:10.1093/bioinformatics/btu620
Knudsen NØ, Nielsen FC, Vinther L, Bertelsen R, Holten-Andersen S, Liberti SE, Hofstra R, Kooi K, Rasmussen LJ (2007) Nuclear localization of human DNA mismatch repair protein exonuclease 1 (hEXO1). Nucleic Acids Res 35:2609–2619. doi:10.1093/nar/gkl1166
Ollila S, Sarantaus L, Kariola R, Chan P, Hampel H, Holinski-Feder E, Macrae F, Kohonen-Corish M, Gerdes AM, Peltomäki P, Mangold E, de la Chapelle A, Greenblatt M, Nyström M (2006) Pathogenicity of MSH2 missense mutations is typically associated with impaired repair capability of the mutated protein. Gastroenterology 131(5):1408–1417. doi:10.1053/j.gastro.2006.08.044
Piñol V, Castells A, Andreu M, Castellví-Bel S, Alenda C, Llor X, Xicola RM, Rodríguez-Moranta F, Payá A, Jover R, Bessa X; Gastrointestinal Oncology Group of the Spanish Gastroenterological Association (2005) Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA 293(16):1986–1994. doi:10.1001/jama.293.16.1986
Petkovic S, Muller S (2015) RNA circularization strategies in vivo and in vitro. Nucleic Acids Res 43:2454–2465
Chen LL (2016) The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol 17:205–211
Ebbesen KK, Kjems J, Hansen TB (2016) Circular RNAs: identification, biogenesis and function. Biochim Biophys Acta 1859:163–168
Li J, Yang J, Zhou P, Le Y, Zhou C, Wang S et al (2015) Circular RNAs in cancer: novel insights into origins, properties, functions and implications. Am J Cancer Res 5:472–480
Chen S, Li T, Zhao Q, Xiao B, Guo J (2017) Using circular RNA hsa_circ_0000190 as a new biomarker in the diagnosis of gastric cancer. Clin Chim Acta 466:167–171
Han D, Li J, Wang H, Su X, Hou J, Gu Y, Qian C, Lin Y, Liu X, Huang M, Li N, Zhou W, Yu Y, Cao X (2017) Circular RNA MTO1 acts as the sponge of miR-9 to suppress hepatocellular carcinoma progression. Hepatology. doi:10.1002/hep.29270
Fu L, Chen Q, Yao T, Li T, Ying S, Hu Y, Guo J (2017) Hsa_circ_0005986 inhibits carcinogenesis by acting as a miR-129-5p sponge and is used as a novel biomarker for hepatocellular carcinoma. Oncotarget 8(27):43878–43888. doi:10.18632/oncotarget.16709
Du WW, Fang L, Yang W, Wu N, Awan FM, Yang Z et al (2017) Induction of tumor apoptosis through a circular RNA enhancing Foxo3 activity. Cell Death Differ 24(2):357–370
Armakola M, Higgins MJ, Figley MD, Barmada SJ, Scarborough EA, Diaz Z et al (2012) Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet 44:1302–1309
Li Z, Huang C, Bao C, Chen L, Lin M, Wang X et al (2015) Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 22:256–264
Kumar L, Shamsuzzama, Haque R, Baghel T, Nazir A. Circular RNAs: the emerging class of non-coding RNAs and their potential role in human neurodegenerative diseases. Mol Neurobiol 1–11
Li P, Chen S, Chen H, Mo X, Li T, Shao Y et al (2015) Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin Chim Acta 444:132–136
Chen J, Li Y, Zheng Q, Bao C, He J, Chen B et al (2016) Circular RNA profile identifies circPVT1 as a proliferative factor and prognostic marker in gastric cancer. Cancer Lett 388:208–219
Acknowledgements
The authors have no funding sources to declare.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Francesca Bianchi and Elena Maccaroni have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Bianchi, F., Maccaroni, E., Belvederesi, L. et al. A germline missense mutation in exon 3 of the MSH2 gene in a Lynch syndrome family: correlation with phenotype and localization assay. Familial Cancer 17, 215–224 (2018). https://doi.org/10.1007/s10689-017-0030-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-017-0030-x