
Abstract
Cotton is one of the most important cash crops around the world, providing natural fiber for the textile industry. Agronomic traits play an important role in the mechanized harvesting of cotton. Plant height (PH) and branch number (BN) are two important traits that could affect plant architecture and ultimately, economic yield of cotton. The quantitative trait locus (QTL) for PH and BN across seven environments was identified with a high-density single nucleotide polymorphism map constructed using recombinant inbred lines from upland cotton 0–153 and sGK9708. A total of 68 QTLs for PH (nine stable) and 64 QTLs for BN (eight stable) were identified. Among these stable QTLs, three (two for PH and one for BN) have been identified in previous studies. Four hundred genes for PH and 624 genes for BN were located on the confidence intervals of these stable QTLs. Among them, 134 for PH and 224 for BN were expressed in at least one tissue of root, stem and leaf. Based on the annotation information, expression pattern and the function validated on the other species, ten genes (six for PH and four for BN) could be considered as potential candidate genes. These results could contribute to understanding the mechanism of PH and branch formation, and to improving the method of cotton breeding for molecular marker-assisted selection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Among the four cultivated Gossypium species, G. hirsutum (upland cotton) accounts for 90–95% of the total cotton fiber production because of its attractive fiber quality and high yield (Rong et al. 2004; Park et al. 2005; Lacape et al. 2009). Improved human living standards are increasing demand for cotton with high-quality traits, creating a challenge for cotton breeders. Plant architecture is one of the most important parts of agronomic traits related to the suitability of mechanical harvest, plant adaptability, plant density, yield and quality (Chen et al. 2019; Su et al. 2018). PH is a major trait affecting plant architecture and serves as a model to analyze dynamic development, which directly determines the biomass and has indirect influence on cotton economic yield. BN has large influences on morphological structure, photosynthetic capacity (Adawy et al. 2013; Guo et al. 2013; He et al. 2014; Shen et al. 2014; Shang et al. 2015, 2016; Mei et al. 2017; Chen et al. 2019; Ma et al. 2018; Su et al. 2018). Most of the plant architecture traits, including PH and BN, are quantitative traits controlled by multiple genes and greatly influenced by environments (Adawy et al. 2013). Therefore, marker-assisted selection (MAS) could be used for improving these traits. Previous research identified QTLs for PH and BN with different kinds of markers and populations (Adawy et al. 2013; Guo et al. 2013; He et al. 2014; Shen et al. 2014; Shang et al. 2015, 2016; Mei et al. 2017; Qi et al. 2017; Ma et al. 2018) such as using simple sequence repeat (SSR) genetic maps (Guo et al. 2013, Shang et al. 2015, 2016; Mei et al. 2017; Ma et al. 2018), amplified fragment length polymorphism (AFLP) and random amplified polymorphic DNA (RAPD) genetic maps (Adawy et al. 2013). Studies that used an SNP genetic map (Qi et al. 2017) did not use the entire upland cotton genome and some QTLs had large CIs. From these, we determined that it is necessary to identify QTLs and potential candidate genes for PH and BN with a high-density map.
In this research, we identified the QTLs for PH and BN across seven environments with a high-density genetic map (Zhang et al. 2016), constructed by an intraspecific RIL population with two parents, 0-153 and sGK9708 (Sun et al. 2012; Jamshed et al. 2016; Zhang et al. 2016, 2017). The stable QTLs were selected and compared with the database cottonQTLdb (Rong et al. 2007; Lacape et al. 2009; Said et al. 2013, 2015a, b; Fang et al. 2014). The genes located on the CI of the stable QTLs were annotated with ontology (GO), Kyoto encyclopedia of genes and genomes (KEGG) and eukaryotic orthologous groups (KOG) database, and the expression pattern of them in root, stem and leaf was analyzed with the RNA-Seq data in Zhang’s research (Zhang et al. 2015a). Combined all these results and the previous studies, the potential candidate genes were identified. This research could provide information about understanding the formation of PH and BN, and employ in marker assisted selection for molecular breeding with improved agronomic traits PH and BN.
Plant materials
An RIL population with 196 individuals was constructed with upland cotton 0–153 and sGK9708 as parents. For the parent 0–153, the mean values of PH and BN in seven environments were 91.14 cm and 13.9 respectively and for the parent sGK9708 were 82.72 cm and 11.1 respectively. F6:8 families were grown in 2007 and the later generations were considered as RILs. The process of developing the population was detailed in Sun’s research (Sun et al. 2012). The phenotypic data of PH and BN were collected across seven environments from 2007 to 2012. The seeds for the seven environments cross 5 years were from F6:8 and the seeds from the last generation were used for next year. The environments included Anyang (AY) of Henan province in 2007, 2008 and 2011, Quzhou (QZ) of Hebei province in 2008, Linqing (LQ) of Shandong province in 2008 and Zhengzhou (ZZ) of Henan province in 2011 and 2012 (Sun et al. 2012; Jamshed et al. 2016; Zhang et al. 2015b, 2016, 2017). In all the seven environments, the phenotypic evaluations of the population were conducted in a completely randomized block design with two replications. In 2007AY and 2008AY, a single row plot was used and each row was 5 m long and 0.8 m wide, the sowing days were April 25 and April 26 respectively; in 2008QZ and 2008LQ, the plastic-membrane covering technique and a wide/narrow row distance planting pattern were used with rows 5 m long and 0.5/0.8 m wide apart, the sowing days were April 27 and April 28 respectively (Sun et al. 2012); In 2011AY, 2011ZZ and 2012ZZ, a single row plot was used and each row was 5 m long and 0.8 m wide, the sowing days were April 26 in the three environments. The plant density was 0.25 m/per plant for every row and the local commercial field management practices were used in each environment.
Collection and statistical analysis of phenotypic data
The phenotype data of PH and BN were collected in the middle of September, and ten plants in each plot were selected in the seven environments. The trait PH was measured from the cotyledonary node to the topping place of the whole plant; the trait BN was counted for all the fruiting branches. The descriptive statistics such as mean value, standard deviation, skewness and kurtosis of PH and BN across the whole population were analyzed with SPSS 20.0 (https://www.ibm.com/analytics/spss-statistics-software). Variance and heritability were analyzed with ICIMAPPING 4.1 (Li et al. 2007, 2015b; Meng et al. 2015). Heritability greater than 40% was considered high heritability; heritability between 20% and 40% was considered medium heritability; and heritability with less than 20% was considered low heritability (Bai et al. 2016; Wang et al. 2018).
QTLs identification for PH and BN
The QTLs were identified based on a high-density genetic map constructed by Zhang et al. (2016) with the phenotype data of seven environments using the composite interval mapping method (CIM) as provided in Windows QTL Cartographer 2.5 (Zeng 1994; Wang et al. 2001). The logarithm of odds (LOD) value for declaring significant QTLs across environments was calculated by a permutation test with the mapping step of 1.0 cM, five control markers, and a significance level of p < 0.05, n = 1000. The QTLs were named as follows: the QTL designations begin with “q” followed by the trait abbreviation, the chromosome number and the QTL serial number (Sun et al. 2012; Zhang et al. 2015b, 2016, 2017; Jamshed et al. 2016).
A positive additive effect indicated that favorable alleles were from parent 0–153 while a negative effect indicated parent sGK9708. The QTLs for the same trait for at least two environments were considered stable when their CIs overlapped (Sun et al. 2012). The stable QTLs were compared with the QTLs in the CottonQTLdb database (Rong et al. 2007; Lacape et al. 2009; Said et al. 2013, 2015a, b; Fang et al. 2014) (http://www2.cottonqtldb.org:8081/). QTLs were newly identified if they did not share or overlap physical CIs with prior studies in the database.
Gene identification and annotation
All the markers that located on the CIs of stable QTLs were compared to the genome of upland cotton (Li et al. 2015a, b; Zhang et al. 2015a) to get their physical position. The genes that located on the physical CIs of the stable QTLs were annotated. The functions of all the genes were forecasted by referring to their corresponding genes in A.thaliana. The genes were also annotated with GO, KEGG and KOG database. We used the go_20160702-seqdb.fasta (http://archive.geneontology.org/latest-lite/) and gene2go (ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/) databases for the GO annotation and the KOG database (ftp://ftp.ncbi.nih.gov/pub/COG/KOG) for the KOG annotation. The sequences of the candidate genes were compared with the sequences in the database using BlastX (e < 10−10) software. KOBAS 3.0 (Xie et al. 2011) (http://kobas.cbi.pku.edu.cn/) software was used for the KEGG annotation.
In-silico RNA-Seq data analysis
RNA-seq data about different tissue of upland cotton (PRJNA248163) were downloaded from the Sequence Read Archive (SRA) of the national center for biotechnology information (NCBI) (https://www.ncbi.nlm.nih.gov/) (Zhang et al. 2015a). First, the data of SRA format were transformed into FASTQ format with the software SRA Toolkit (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software) (–split-3), then the software Bowtie 2 (Langmead and Salzberg 2012) was used to construct the library and the software Tophat 2 (Kim et al. 2013) was used to compared the FASTQ format reads to the upland cotton genome (Li et al. 2015a; Zhang et al. 2015a). The software Cufflinks were used to calculate the fragments per kilobase million (FPKM) value (Trapnell et al. 2010). The genes with FPKM value more than 10 in at least one tissue could be considered as expression genes (Zhang et al. 2019). The heatmap was drawn with the value of log2(FPKM+1) by the software Pheatmap in the R package.
Results and discussion
The descriptive statistics and heritability analysis of PH and BN
The descriptive statistics of PH and BN for parents and the RIL population in seven environments are summarized in Table 1. All traits showed approximately normal distributions with an absolute skewness value of less than one and were characterized by transgressive segregations with respect to their parental performances during the evaluations (Table 1). Heritability for PH and BN was calculated across seven environments. For PH, heritability ranged from 0.34 to 0.62. Results indicated high heritability in five of the seven environments and showed medium heritability in the remaining two environments. For BN, the heritability ranged from 0.23 to 0.42. Results indicated high heritability in two of the seven environments and showed medium heritability in the remaining five environments. The variance analysis and heritability analysis results are shown in Table S1.
As the traits PH and BN were both quantitative traits and influenced greatly by environment, so identify QTLs for these two traits across multiple environments and selected the stable ones could be more accurately. Under the background of SNP genetic map widely used in QTL identification, in previous studies, most of the studies focused on QTLs for PH and BN using SSR map (Guo et al. 2013; He et al. 2014; Shang et al. 2015, 2016; Mei et al. 2017; Ma et al. 2018). Among the few reports that used SNP map, Qi’s research used only one environment in F2 population and one environment in F2:3 population (Qi et al. 2017) while in our research, total seven environments were used to identify QTLs. These could contribute to future research such as QTL identification, fine mapping, function genes cloning and also provide some reference for discovering the mechanism of PH and BN formation.
QTL identification and congruence analysis with previous reports
For PH, there were 68 QTLs on 22 of 26 chromosomes except chromosome 2, 6, 11, and 24 (Table S2 and Table 2 and Fig. 1). Among the results, nine stable QTLs could be detected in at least two environments. The qPH-chr16-1 was detected in five environments, located on the CI of 100.4–104.6 cM, and explained 4.38–8.96% of the observed phenotypic variation (PV) with negative additive effects; the QTLs qPH-chr5-5, qPH-chr7-1 and qPH-chr12-3 were detected in three environments, located on CIs of 185.9 cM-194.3 cM of chr.5, 13.7 cM–21.10 cM of chr.7 and 29.5 cM–32.4 cM of chr.12, and explained 5.67–9.64%, 5.06–6.68% and 7.07–11.13% of the observed PV, respectively, all with negative additive effects. The QTLs qPH-chr1-4, qPH-chr1-6, qPH-chr7-5, qPH-chr17-1 and qPH-chr26-1 were detected in two environments, located on the CIs of 74.5–76.2 cM of chr.1, 99.3–102.00 cM of chr.1, 119.7–121.4 cM of chr.7, 6.2–13.2 cM of chr.17 and 56.3–61.5 cM of chr.26, and explained 5.04–6.02%, 4.66–6.23%, 3.81–7.68%, 4.51–4.80% and 5.56–5.67% of the observed PV, respectively, all with positive additive effects.

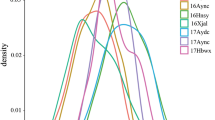
The variation trend of the LOD value along with the genetic distance of stable QTLs for PH and BN in specific one of the related environments. a The variation trend of the LOD value along with the genetic distance of stable QTLs for PH in specific one of the related environments. b The variation trend of the LOD value along with the genetic distance of stable QTLs for BN in specific one of the related environments
For BN, there were 64 QTLs located on all 26 chromosomes (Table S2, Table 2 and Fig. 1). Among them, eight stable QTLs were detected in two environments. The QTLs qBN-chr2-2, qBN-chr4-2, qBN-chr8-1, qBN-chr15-1, qBN-chr18-2, qBN-chr19-1, qBN-chr19-2 and qBN-chr25-3 were detected in two environments, located on the CIs of 12.9–19.2 cM of chr2, 95.3–97.0 cM of chr4, 30.4–37.1 cM of chr8, 0.0–2.9 cM of chr15, 132.5–135.9 cM of chr18, 0.0–6.1 cM of chr19, 16.9–22.0 cM of chr19 and 89.5–91.3 cM of chr25, and explained 6.19–6.43%, 6.45–8.74%, 4.67–5.47%, 5.24–7.17%, 4.81–6.42%, 5.72–6.40%, 5.74–6.32% and 5.21–5.97% of the observed PV, respectively. The additive effects for qBN-chr2-2, qBN-chr4-2, qBN-chr8-1, qBN-chr19-2 and qBN-chr25-3 were negative, while additive effects for qBN-chr15-1, qBN-chr18-2 and qBN-chr19-1 were positive (Table S2; Table 2).
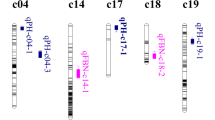
To discover whether the stable QTLs in our study were novel or had been identified in previous studies, we compared the stable QTLs with those in the cottonQTLdb (Rong et al. 2007; Lacape et al. 2009; Said et al. 2013, 2015a, b; Fang et al. 2014). As in this database, all the markers were SSR markers and restriction fragment length polymorphism (RFLP) markers. Therefore meta-analysis could not be used as no common markers could be found between our SNP map and the SSR and RFLP maps in the database. The QTLs shared the same or overlapped physical CIs in the database and our results were common QTLs. For PH, the QTLs in the database were distributed on the 23 chromosomes except chromosome 4, 12, and 18. In our research, we also identified one stable QTL for PH on chromosome 12. For BN, the QTLs in the database were distributed on 18 chromosomes except chromosome 3, 4, 7, 8, 15, 19, 20 and 21. In our research, we also identified stable QTLs on chromosome 4, 7, 8, 15 and 19. Comparing the physical position between the QTLs in our research and the QTLs in the database, two QTLs for PH (qPH-chr1-4 and qPH-chr1-6) and one QTL for BN (qBN-chr25-3) should be considered as common QTLs. The other seven QTLs for PH and eight QTLs for BN were newly identified (Fig. 2). Previous studies identified QTLs with an SNP map, but most of the results focused on fiber quality and yield traits (Hulsse-Kemp et al. 2015; Wang et al. 2015a, b; Li et al. 2016; Tan et al. 2018). For the trait PH and BN, there were only a few researches. In Qi’s research, due to the unknown physical position of their SNP markers, we could not compared the results with ours, but that research have not identified any stable QTLs, each QTLs could be detected in only one generation and one environment (Qi et al. 2017).

Physical position of QTLs both could be identified in the database and in our research. a The physical position of qPH-chr1-4 and qPH-chr1-6 in the database and in our research. b The physical position of qPH-chr5-5 in the database and in our research. c The physical position of qBN-chr25-3 in the database and in our research
In our research, we increased the density of the markers and shortened the CIs of the QTLs in the database. The QTL on chromosome 1 for PH in the database harbored five SSR markers, four were located on the physical CIs from 86.31 to 98.15 MB and the other one located on 44.06 MB. In our research, the QTLs qPH-chr-4 and qPH-chr-6 were located on the physical CIs from 85.27 to 85.40 MB and 94.47 to 94.70 MB. One QTL with large CIs in the database were divided into two QTLs with smaller CIs. The QTL on chromosome 5 for PH in the database harbored three SSR markers and were located on the physical CI from 86.77 to 91.53 MB. In our research, the qPH-chr5-5 was located on the physical CI from 90.37 to 90.71 MB, much smaller than in the database. The QTL on chromosome 25 for BN in the database harbored 11 SSR markers and were located on the physical CI from 5.18 to 34.62 MB. In our research, the qBN-chr25-3 was located on the physical CI from 11.04 to 12.61 MB, much smaller than in the database. For the previously identified QTLs, we narrowed the CIs from the database. For the new data, we identified five important QTLs, including qPH-chr16-1, qPH-chr5-5 and qPH-chr12-3 for PH, and qBN-chr4-2, and qBN-chr15-1 for BN, which were detected in multiple environments and explained more than 7% of the observed PV. These results could promote further research into the mechanisms that control PH and BN, and could also provide a basis for MAS (Fig. 2).
Gene identification, annotation and expression pattern analysis
A total number of 1024 genes (400 for PH and 624 for BN) were located on the CIs of stable QTLs and used to do the next step of analysis. For PH, the qPH-chr26-1 harbored the most 248 genes, while the qPH-chr1-4 harbored the least one gene (Table 3). For BN, the qBN-chr15-1 harbored the most 282 genes, while the qBN-chr4-2 harbored the least ten genes. The qBN-chr2-2, qBN-chr18-2 and qBN-chr19-1 harbored no genes because of the small physical CIs (Table 3). The function of all the genes for PH and BN were forecasted by identifying its corresponding genes in A. thaliana (Table S3). The genes were also annotated with the database GO, KEGG and KOG. For GO annotation, the GO term “biological process” in the category “biological process”, the GO term “nucleus” in the category “cellular component” and “molecular function” in the category “molecular function” harbored the most number of genes for both traits. For PH, the three GO terms harbored 61, 120, and 58 genes, respectively. For BN, the three GO terms harbored 97, 159 and 91 genes, respectively (Table S4, Fig. S1). For KEGG annotation, the KEGG pathway “metabolic pathways”, “biosynthesis of secondary metabolites” and “ribosome” harbored the most genes for both traits. For PH, these three pathways harbored 20, 16 and seven genes, respectively. For BN, these three pathways harbored 62, 40 and 12 genes, respectively (Table S5, Fig. S1). For KOG annotation, the KOG basket “signal transduction mechanisms” and “transcription” had the most number of genes for both traits. For PH, the two baskets harbored 41 and 29 genes, respectively. For BN, the two KOG baskets harbored 47 and 28 genes, respectively. Some genes have no clear KOG annotation information, as they were located on the baskets “general function prediction only” and “function unknown”. For PH, there were 51 and 18 genes located on these two baskets, respectively. For BN, there were 98 and 30 genes located on these two baskets, respectively (Table S6, Fig. S1).
For PH, there were 134 genes that expressed in at least one of the three tissues (root, stem and leaf). Among them, 60 were expressed in all the three tissues, 15 in stem and leaf, two in root and stem; 16 in root and leaf, 14 only in root, 16 only in stem, 134 only in leaf. For BN, there were 224 genes that expressed in at least one of the three tissues. Among them, 109 were expressed in all the three tissues; 22 in stem and leaf, nine in root and stem, 28 in root and leaf, 18 only in root, 25 only in stem, 13 only in leaf (Table S7 and Fig. S2).
Among the 134 genes for PH and 224 for BN, six for PH and four for BN have been validated in the previous researches. For PH, the gene Gh_A05G3508 is related to “cytochrome P450”which takes part in the important biosynthesis pathways, where mutations in the gene may block brassinosteroid biosynthesis, thereby creating dwarf plants (Yang et al. 2014; Qi et al. 2017; Wu et al. 2016); the gene Gh_D07G0671 belongs to MADS-box family which is involved in cellular differentiation and floral determination (Hempel et al. 1997; Gu et al. 1998; Su et al. 2018); the genes Gh_D12G1734, Gh_D03G0226 and Gh_A05G3499 belong to WRKY family which help plants to adapt through physiological and morphological changes and control flowering time and plant height (Cai et al. 2014; Gu et al. 2018); The gene Gh_D12G1644 belongs to TCP family which directly bind the promoters of core cell-cycle genes in Arabidopsis inflorescence and shoot apices (Martı´n-Trillo et al. 2010; Davie`re et al. 2014). For BN, the gene Gh_A08G0299 belongs to MYB family which is the key regulator of early steps during shoot branching process (Schmitz and Theres 2005; Song et al. 2011; Yang et al. 2018).The gene Gh_D05G1192 was annotated with the function of “AMP deaminase, putative/myoadenylate deaminase, putative”; the two genes Gh_D01G2007 and Gh_D01G2145 were annotated with the function of “Transducin/WD40 repeat-like superfamily protein” in Arabidopsis (Bajaj et al. 2016). These ten genes were considered as potential candidate genes, which could provide information of understanding the mechanism of the formation of PH and BN, and also help breeders to improve the PH and BN (Tables S3–S6).
References
Adawy SS, Diab AA, Atia MA, Hussein EH (2013) Construction of genetic linkage map with chromosomal assignment and quantitative trait loci associated with some important agronomic traits in cotton. GM Crops Food 4:36–49
Bai JY, Yang YB, Huang Y, Qi YX, Li GL (2016) The effects of traits heritability on the efficiency of marker assisted introgression. Jiangsu Agric Sci 44:35–37
Bajaj D, Upadhyaya HD, Das S, Kumar B, Gowda CLL, Sharma S, Tyagi AK, Sk Parida (2016) Identification of candidate genes for dissecting complex branch number trait in chickpea. Plant Sci 245:61–70
Cai YH, Chen XJ, Xie K, Xing QK, Wu YW, Li J, Du CH, Sun ZX, Guo ZJ (2014) Dlf1, a WRKY transcription factor, is involved in the control of flowering time and plant height in rice. PLoS ONE 9:e102529
Chen W, Yao JB, Li Y, Zhao LJ, Liu J, Guo Y, Wang JY, Li Y, Liu ZY, Lu YJ, Zhang YS (2019) Nulliplex-branch, a TERMINAL FLOWER 1 ortholog controls plant growth habit in cotton. Theor Appl Genet 132:97–112
Davie`re J, Wild M, Regnault T, Baumberger N, Eisler H, Genschik P, Achard P (2014) Class I TCP-DELLA interactions in inflorescence shoot apex determine plant height. Curr Biol 24:1923–1928
Fang DD, Jenkins JN, Deng DD, McCarty JC, Li P, Wu JX (2014) Quantitative trait loci analysis of fiber quality traits using a random-mated recombinant inbred population in upland cotton (Gossypium hirsutum L.). BMC Genom 15:397
Gu Q, Ferrándiz C, Yanofsky MF, Martienssen R (1998) The FRUITFULL MADS-box gene mediates cell differentiation during Arabidopsis fruit development. Development 125:1509–1517
Gu LJ, Wei HL, Wang HT, Su JJ, Yu SX (2018) Characterization and functional analysis of GhWRKY42, a group IId WRKY gene, in upland cotton (Gossypium hirsutum L.). BMC Genet 19:48
Guo X, Guo YP, Ma J, Wang F, Sun MZ, Gui LJ, Zhou JJ, Song XL, Sun XZ, Zhang TZ (2013) Mapping heterotic loci for yield and agronomic traits using chromosome segment introgression lines in cotton. J Integr Plant Biol 55:759–774
He R, Shi YZ, Zhang JF, Liang Y, Zhang BC, Li JW, Wang T, Gong JW, Liu AY, Shang HH, Gong WK, Bai AC, Yuan YL (2014) QTL mapping for plant height using chromosome segment substitution lines in upland cotton. Acta Agron Sin 40:457–465
Hempel FD, Weigel D, Mandel MA, Ditta G, Zambryski PC, FeldmanL J, Yanofsky MF (1997) Floral determination and expression of floral regulatory genes in Arabidopsis. Development 124:3845–3853
Hulsse-Kemp AM, Lemm J, Plieske J, Ashra H, Burrarapu R, Fang DD, Frelichowski J, Giband M, Hague S, Hinze L, Kochan KJ, Riggs PK, Schef JA, Udall JA, Ulloa M, Wang SS, Zhu QH, Bag SK, Bhardwaj A, Burke JJ, Byers RL, Claverie M, Gore M, Harker DB, Lslam MS, Jenkins JN, Jones DC, Lacape JM, Liewellyn DJ, Percy RG, Pepper AE, Poland JA, Rai KM, Sawant SV, Singh SK, Spriggs A, Taylor JM, Wang F, Yourstone SM, Zhang XT, Lawley CT, Ganal MW, Deynze AV, Wilson IW, Stelly DM (2015) Development of a 63 K SNP array for cotton and high-density mapping of intraspecific and interspecific populations of Gossypium spp. G3 Genes Genomes Genet 5:1187–1209
Jamshed M, Jia F, Gong JW, Plalanga KK, Shi ZY, Li JW, Shang HH, Liu AY, Chen TT, Zhang Z, Cai J, Ge Q, Liu Z, Lu QW, Deng XY, Tan YN, Rashid HO, Sarfrza Z, Hassan M, Gong WK, Yuan YL (2016) Identification of stable quantitative trait loci (QTLs) for fiber quality traits across multiple environments in Gossypium hirsutum recombinant inbred line population. BMC Genom 17:197
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36
Lacape JM, Jacobs J, Arioli T, Derijcker R, Forestier-Chiron N, Llewellyn D, Jean J, Thomas E, Viot C (2009) A new interspecific, Gossypium hirsutum × G. barbadense, RIL population: towards a unified consensus linkage map of tetraploid cotton. Theor Appl Genet 119:281–292
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with bowtie 2. Nat Methods 9:357–359
Li HH, Ye GY, Wang JK (2007) A modified algorithm for the improvement of composite interval mapping. Genetics 175:361–374
Li FG, Fang GY, Lu CR, Xiao GH, Zou CS, Kohel RJ, Ma ZY, Shang HH, Ma XF, Wu JY, Liang XM, Huang G, Percy RG, Liu K, Yang WH, Chen WB, Du XM, Shi CC, Yuan YL, Ye WW, Liu X, Zhang XY, Liu WQ, Wei HL, Wei SJ, Huang GD, Zhang XL, Zhu SJ, Sun FM, Wang XF, Liang J, Wang JH, He Q, Huang LH, Wang J, Cui JJ, Song GL, Wang KB, Xu X, Yu JZ, Zhu YX, Yu SX (2015a) Genome sequence of cultivated upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol 33:524–530
Li SS, Wang JK, Zhang LY (2015b) Inclusive composite interval mapping of QTL by environment interactions in biparental population. PLoS ONE 10:132414
Li C, Dong YT, Zhao TL, Li C, Yu E, Mei L, Daud MK, He QL, Chen JH, Zhu SJ (2016) Genome-wide SNP linkage mapping and QTL analysis for fiber 1uality and yield traits in the upland cotton recombinant inbred lines population. Front Plant Sci 7:1356
Ma LL, Ijaz B, Wang YM, Hua JP (2018) Dynamic QTL analysis and validation for plant height using maternal and paternal backcrossing populations in Upland cotton. Euphytica 214:167
Martı´n-Trillo M, Cubas P (2010) TCP genes: a family snapshot ten years later. Trends Plant Sci 15:31–39
Mei Y, Yu J, Xue A, Fan S, Song M, Pang C, Pei W, Yu S, Zhu J, Zhang J (2017) Dissecting genetic network of fruit branch traits in upland cotton by association mapping using SSR markers. PLoS ONE 12(1):e0162815
Meng L, Li HH, Zhang LY, Wang JK (2015) QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3:269–283
Park YH, Alabady MS, Ulloa M, Sickler B, Wilkin TA, Yu J, Stelly DM, Kohel RJ, El-Shihy OM, Cantrell RG (2005) Genetic mapping of new cotton fiber loci using EST-derived microsatellites in an interspecific recombinant inbred line cotton population. Mol Gen Genom 274:428–441
Qi HK, Wang N, Qiao WQ, Xu QH, Zhou H, Shi JB, Yan GT, Huang Q (2017) Construction of a high-density genetic map using genotyping by sequencing (GBS) for quantitative trait loci (QTL) analysis of three plant morphological traits in upland cotton (Gossypium hirsutum L.). Euphytica 213:83
Rong JK, Abbey C, Browers JE, Brubaker CL, Cheng C, Chee PW, Delmonte TA, Ding XL, Garza JJ, Marler BS, Park CH, Pierce GJ, Rain KM, Rastogi VK, Schulze SR, Trolinder NL, Wendel JF, Wilkins TA, Williams-Coplin TD, Wing RA, Wright RJ, Zhao XP, Zhu LH, Paterson AH (2004) A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium). Genetics 166:389–417
Rong JK, Feltus FA, Waghmare VN, Pierce GJ, Chee PW, Graye X, Sarange Y, Wright RJ, Wilkins TA, May L, Smith W, Gannaway JR, Wendel JF, Paterson AH (2007) Meta-analysis of polyploid cotton QTL shows unequal contributions of sub genomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics 176:2577–2588
Said JI, Lin ZX, Zhang XL, Song MZ, Zhang JF (2013) A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom 14:776
Said JI, Knapka JA, Song MZ, Zhang JF (2015a) Cotton QTLdb: a cotton QTL database for QTL analysis, visualization, and comparison between Gossypium hirsutum and G. hirsutum × G. barbadense populations. Mol Genet Genom 290:1615–1625
Said JI, Song MZ, Wang HT, Lin ZX, Zhang XL, Fang DD, Zhang JF (2015b) A comparative meta-analysis of QTL between intraspecific Gossypium hirsutum and interspecific G. hirsutum × G. barbadense populations. Mol Genet Genom 290:1003–1025
Schmitz G, Theres K (2005) Shoot and inflorescence branching. Curr Opin Plant Biol 8:506–511
Shang LG, Liu F, Wang YM, Abduweli A, Cai SH, Wang KB, Hua JP (2015) Dynamic QTL mapping for plant height in upland cotton (Gossypium hirsutum). Plant Breed 134:703–712
Shang LG, Ma LL, Wang YM, Su Y, Wang XC, Li YH, Abduweli A, CaiSH Liu F, Wang KB, Hua JP (2016) Main effect QTL with dominance determines heterosis for dynamic plant height in upland cotton. G3: genes. Genome Genet 6:3373–3379
Shen GJ, Zhan W, Chen HX, Xing YZ (2014) Dominance and epistasis are the main contributors to heterosis for plant height in rice. Plant Sci 215–216:11–19
Song SS, Qi TC, Huang H, Ren QC, Wu DW, Chang CQ, Peng W, Liu YL, Peng JR, Xie DX (2011) The jasmonate-ZIM domain proteins interact with the R2R3-MYB transcription factors MYB21 and MYB24 to affect jasmonate-regulated stamen development in Arabidopsis. Plant Cell 23:1000–1013
Su JJ, Li LB, Zhang C, Wang CX, Gu LJ, Wang HT, Wei HL, Liu QB, Huang L, Yu SX (2018) Genome-wide association study identified genetic variation sand candidate genes for plant architecture component traits in Chinese upland cotton. Theor Appl Genet 131:1299–1314
Sun FD, Zhang JH, Wang SF, Gong WK, Shi YZ, Liu AY, Li JW, Gong JW, Shang HH, Yuan YL (2012) QTL mapping for fiber quality traits across multiple generations and environments in upland cotton. Mol Breed 30:569–582
Tan ZY, Zhang ZQ, Sun XJ, Li QQ, Sun Y, Yang P, Wang WW, Liu XY, Chen CL, Liu DX, Teng ZH, Guo K, Zhang J, Liu DJ, Zhang ZS (2018) Genetic map construction and fiber 1uality QTL mapping using the cottonSNP80 K array in upland cotton. Front Plant Sci 9:225
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515
Wang S, Basten CJ, Zeng ZB (2001) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC.http://statgen.ncsu.edu/qtlcart/WQTLCart.htm. Accessed 1 Feb 2013
Wang HT, Jin X, Zhang BB, Shen C, Li ZX- (2015a) Enrichment of an intraspecific genetic map of upland cotton by developing markers using parental RAD sequencing. DNA Res 22:147–160
Wang YK, Ning ZY, Hu Y, Chen JD, Zhao R, Chen H, Ai NJ, Guo WZ, Zhang TZ (2015b) Molecular mapping of restriction-site associated DNA markers in allotetraploid upland cotton. PLoS ONE 10:e0124781
Wang CT, Wang XZ, Wu Q, Wang ZW, Tang YY, Du ZB, Liu F, Li Q (2018) Sensory quality of fresh green peanuts: dominance analysis, selection of elite lines and heritability. J Peanut Sci 47:71–74
Wu YZ, Fu TC, Zhao SS, Gu P, Zhu ZF, Sun CQ, Tan LB (2016) CLUSTERED PRIMARY BRANCH 1, a new allele of DWARF11, controls architecture and seed size in rice. Plant Biotechnol J 1:377–386
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong J, Gao G, Li CY, Wei L (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39:316–322
Yang ZR, Zhang CJ, Yang XJ, Liu K, Wu ZX, Zhang XY, Zheng W, Xun QQ, Liu CL, Lu LL, Yang ZE, Qian YY, Xu ZZ, Li CF, Li J, Li FG (2014) PAG1, a cotton brassinosteroid catabolism gene, modulates fiber elongation. New Phytol 203:437–448
Yang H, Xue Q, Zhang ZZ, Du JY, Yu DY, Huang F (2018) GmMYB181, a Soybean R2R3-MYB protein, increases branch number in transgenic Arabidopsis. Front Plant Sci 9:1027
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zhang TZ, Hu Y, Jiang W, Fang L, Guan X, Chen J, Zhang J, Saski CA, Scheffler BE, Stelly DM, Hulse-Kemp AM, Wan Q, Liu B, Liu C, Wang S, Pan M, Wang Y, Wang D, Ye W, Chang L, Zhang W, Song Q, Kirkbride RC, Chen X, Dennis E, Llewellyn DJ, Peterson DG, Thaxton P, Jones DC, Wang Q, Xu X, Zhang H, Wu H, Zhou L, Mei G, Chen S, Tian Y, Xiang D, Li X, Ding J, Zuo Q, Tao L, Liu Y, Li J, Lin Y, Hui Y, Cao Z, Cai C, Zhu X, Jiang Z, Zhou B, Guo W, Li R, Chen ZJ (2015a) Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides are source for fiber improvement. Nat Biotechnol 33:531–537
Zhang Z, Li JW, Muhammad J, Cai J, Jia F, Shi YZ, Gong JW, Shang HH, Liu AY, Chen TT, Ge Q, Palanga KK, Lu QW, Deng XY, Tan YN, Li W, Sun LY, Gong WK, Yuan YL (2015b) High resolution consensus mapping of quantitative trait loci for fiber strength, length and micronaire on chromosome 25 of the upland cotton (Gossypium hirsutum L.). PLoS ONE 10(8):e0135430
Zhang Z, Shang HH, Shi YZ, Huang L, Li JW, Ge Q, Gong JW, Liu AY, Chen TT, Wang D, Wang YL, Palanga KK, Muhammad J, Li WJ, Lu QW, Deng XY, Tan YN, Song WW, Cai J, Li PT, Rashid H, Gong WK, Yuan YL (2016) Construction of a high-density genetic map by specific locus amplified fragment sequencing (SLAF-seq) and its application to quantitative trait loci (QTL) analysis for boll weight in upland cotton (Gossypium hirsutum). BMC Plant Biol 16:79
Zhang Z, Ge Q, Liu AY, Li JW, Gong JW, Shang HH, Shi YZ, Chen TT, Wang YL, Palanga KK, Muhammad J, Lu QW, Deng XY, Tan YN, Liu RX, Zou XY, Rashid H, Iqbal MS, Gong WK, Yuan YL (2017) Construction of a high-density genetic map and its application to QTL identification for fiber strength in upland cotton. Crop Sci 57:774–788
Zhang Z, Zou X, Huang Z, Fan S, Qun G, Liu A, Gong J, Li J, Gong W, Shi Y, Fan L, Zhang Z, Liu R, Jiang X, Lei K, Shang H, Xu A, Yuan Y (2019) Genome-wide identification and analysis of the evolution and expression patterns of the GATA transcription factors in three species of Gossypium genus. Gene 680:72–83
Acknowledgements
This work was funded by The Natural Science Foundation of China (31471538, and 31371668) and the central level of the scientific research institutes for basic R & D special fund business (161012018017). We thank Elizabeth Martell, MSc, from LiwenBianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, Z., Liu, A., Huang, Z. et al. Quantitative trait locus mapping for plant height and branch number in an upland cotton recombinant inbred line with an SNP-based high-density genetic map. Euphytica 215, 110 (2019). https://doi.org/10.1007/s10681-019-2428-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-019-2428-z