
Abstract
Segregation distortion analysis in plant breeding is highly important because deviation factors are occasionally serious barriers for introgression of genes of interest into cultivated species. Also, the identification of major QTLs by high linkage disequilibrium is highly useful for breeders in order for crop improvement programs and the further fine mapping of particular genes. Consequently, simultaneous consideration of segregation distortion and linkage disequilibrium leads to developing fertile hybrids and normal inheritance of agronomically favorable genes. Hence, the goal of this study was to investigate genomic regions known to contain fragrance quantitative trait loci (QTLs) in terms of segregation distortion and linkage disequilibrium pattern, and to seek for a possible relationship between them using codominant markers on chromosomes 3, 4, and 8 in a set containing 189 F2 progenies. For the first time, we discovered the presence and extent of segregation distortion and linkage disequilibrium across the fragrance chromosomal areas of rice. The high frequency of skewed allelic segregations in the chromosomes 3 and 4 supports reports of possible gametic selection (ga) and hybrid sterility (S) genes. Finally, the highest linkage disequilibrium values, the lowest recombination frequency, and the shortest distances were observed in the chromosomal regions without segregation distortion. High linkage disequilibrium correlations between markers at short distances may show that such markers can maintain high linkage disequilibrium with linked QTLs and are influential for both MAS and QTL mapping analysis. Moreover, an indirect relationship between segregation distortion and linkage disequilibrium was detected, and thus linkage disequilibrium maps were introduced as useful diagnostic tools for segregation distortion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Departure from the expected Mendelian segregation ratio of alleles at a locus, and consequently at other loci linked to it, will occur when gametic or zygotic fitness decreases (Alheit et al. 2011; Baumbach et al. 2012). In addition to rice (Xu et al. 1997), these causes of segregation distortion have been identified in other species, such as tomato (Paterson et al. 1988), alfalfa (Brummer et al. 1993), maize (Lu et al. 2002), triticale (Alheit et al. 2011), and Citrus (Ollitrault et al. 2012). Many factors influence the rate and chromosomal location of segregation aberration, for instance, mapping population, genetic transmission, gametophytic selection (ga) genes, and hybrid breakdown genes (Alheit et al. 2011; Baumbach et al. 2012; Kumar et al. 2007; Matsushita et al. 2003; Wu et al. 2010). Additionally, environmental agents, non homologous recombination (Knox and Ellis 2002), epistasis (Carlborg et al. 2006), and chromosomal structures like ones affecting movement on spindle fibers during meiosis (Lyttle 1991) have been reported to cause non-Mendelian segregation among alleles. Gene mutation in meiotic division or early phases of reproductive cell development can also impact allele frequency in the gametes (Baumbach et al. 2012).
The segregation distortion observed in different populations and in different chromosomal regions within the same population may have different causes (Alheit et al. 2011). Thus far, F2 populations have generated numerous genetic linkage maps in plant for the sake of easy establishment in a short time and been beneficial for genetic linkage map construction. The advancement of high-density molecular linkage maps provides opportunity to survey the entire genome for loci presenting distorted segregation (Zhao et al. 2006). Segregation distortion analysis has been used to illuminate the significant systems differentiating and isolating species, and is also vital for plant breeding (Matsushita et al. 2003). Distorted markers influence the estimation of genetic intervals and the markers order on a same chromosome. Although some algorithms are used to modify linkage distance estimated based on expected segregation distortion, there are no clear principles to figure out the pseudo-linkages or insufficient linkages of genotypes to phenotypes. However, the adjusted linkage maps can be used for further quantitative trait loci (QTLs) or segregation distortion locus analyses (Zhu et al. 2007). Nevertheless, if distorted markers are managed appropriately, not only their effects on genetic map construction and QTL mapping will decrease (Alheit et al. 2011; Xu and Hu 2009), but also can be fruitful for QTL mapping (Alheit et al. 2011; Xu 2008).
The terms linkage disequilibrium and association mapping have been mostly used interchangeably in literature. However, association mapping considers high association of a molecular marker with a trait, whereas linkage disequilibrium refers to non-random association between two markers or two genes/QTLs or between a gene/QTL and a marker locus. Therefore, association mapping is among one of the applications of linkage disequilibrium. Statistically, association is covariance of a marker polymorphism and a trait of interest, whereas linkage disequilibrium refers to covariance of polymorphisms represented by two molecular markers/genes (Gupta et al. 2005). The extent of linkage disequilibrium is an interesting topic (Zhao et al. 2007) and can be used for different goals such as marker–marker association (non-random association between two markers), marker–trait association (without a mapping population) pursued via marker assisted selection (MAS), marker–QTL/gene association, and genetic diversity in many populations as well as germplasm collections. Linkage disequilibrium can be derived from selection, physical linkage, population structure, and drift (Gupta et al. 2005). Rice, predominantly autogamous, is expected to result in extensive linkage disequilibrium, maybe even genome-wide linkage disequilibrium. The understanding of linkage disequilibrium in rice can help in assessing the effectiveness of linkage disequilibrium mapping in other autogamous species (Garris et al. 2003).
In order to share genomic and genetic knowledge of species, comparative genetic mapping can be an influential approach (Feuillet and Keller 2002). Simple sequence repeat (SSR) marker development and linkage map construction were carried out in order to accelerate the progress of molecular genetics in rice. To expand the knowledge across rice species, we compared the genome structure of our population with that of another rice population (http://www.gramene.org).
Fragrance in rice is known as a valuable characteristic determining consumer preference and the premium price in world trade. Although the inheritance of rice fragrance has been reported to be controlled by one or two or three recessive or dominant genes or by QTLs (showing the complicated genetic monitoring of this trait), several crucial obstacles and ambiguities are still observable in genetic and genomic analysis of fragrant rice. These obstacles can be counted as detection of effective fragrance QTLs, location of fragrance genes, interacting QTLs, and their impact on this important feature (Golestan Hashemi et al. 2013). Besides these ambiguities, there is no report regarding segregation distortion and linkage disequilibrium across the known fragrance genomic regions. Initially, we performed a preliminary aroma QTL mapping and found only one region with high LOD score on chromosome 8 with major effect. Additionally, by a survey on literature, we found that so far chromosomes 3 (Amarawathi et al. 2008) and 4 (Amarawathi et al. 2008; Lorieux et al. 1996) with minor effects, and chromosome 8 (Bradbury 2009; Sun et al. 2008; Yi et al. 2009) with major effect have been introduced as carriers for aroma genes. Consequently, our result was mostly in line with the findings of the previous studies. Hence, in the present research, we only focused on the important chromosomes (including chromosomes 3 and 4) conferring fragrance trait to evaluate the frequency and extent of segregation distortion and linkage disequilibrium, and a possible relationship between them in our segregating population.
Materials and methods
Population development and genomic DNA extraction
The present research is based on 189 F2 progenies developed from crossing between Malaysian non-aromatic cultivar, ‘MR84’ and the Malaysian aromatic rice cultivar, ‘MRQ74’. F2 individuals were grown in normal rice growing season at Malaysian Agriculture Research and Development Institute (MARDI) in an augmented field design with parental lines. Fresh leaf tissues were harvested from single seedling and then taken to the laboratory in order to be frozen in liquid nitrogen. High-quality genomic DNA was extracted from 20 to 25 mg of frozen leaf tissues based on a developed cetyltrimethyl ammonium bromide (CTAB) approach (Gawel and Jarret 1991) and adapted to a concentration of ca. 70 ng/l for analysis of markers.
Molecular marker analysis and segregation distortion
One hundred twelve pairs of SSR primers (First BASE Laboratories Sdn Bhd Co., Ltd, Malaysia) covering chromosomes 3, 4, and 8 of rice were selected from the rice molecular linkage map and microsatellite database published (http://www.gramene.org) (McCouch et al. 2002; Temnykh et al. 2000) for comparative polymorphic evaluation among the parental inbred lines and two randomly selected F1 individuals.
In order to genotype the F2 progenies with polymorphic primers, a touchdown polymerase chain reactions (PCR) protocol was performed (Bradbury et al. 2005). For all progenies, the loci were amplified in PCR reactions in a volume of 15 μl including genomic DNA as template, 0.1 µM forward and reverse primer, 80 µM deoxyribonucleotide triphosphate (dNTPs), 2 mM MgCl2, and 0.5 U of DNA Taq polymerase in a T100-Thermal Cycler (BIO-RAD, USA). Then, differences in restriction banding pattern were resolved on a 3 % MetaPhore gel.
A cluster analysis was performed to group markers based on their genotypes using R statistical software (R Core Team 2011). Based on the expected allelic ratio in the F2 population (1A:2H:1B), the segregation fit of each locus was investigated according to the P values gained (P < 0.05) by a Chi square test using the R/qtl software (Broman et al. 2003). The result of Chi square test and bi-parent genotyping determined the direction and reason of distortion.
Construction of linkage map
In order to construct the genetic linkage map, based on the parents’ scores, genotype codes were recorded from SSR markers polymorphism in the population. A pre-selection regarding the segregation ratio and genotyping errors was carried out for quality filtering, as markers significantly (P < 1e − 10) deviated from the expected 1:2:1 ratio in a Chi square test, and with a genotyping error LOD = 2 would be excluded from further investigation. The genetic linkage map was developed with the R/qtl package (Broman et al. 2003). Markers were classified in linkage groups that apply the parameter of the independence LOD with the arguments min.LOD = 4 and max.rf = 0.35, as a default. During calculating of the individual map, estimation of recombination frequencies and the locus order within chromosomes were founded employing the provided maximum likelihood algorithm. The ‘orderMarkers’ function was used for arranging an initial order. Then, with the argument use.ripple = TRUE, the ‘ripple’ function was employed to investigate all possible orders to improve marker order. The Kosambi’s mapping function was used for calculating map distances in centiMorgans (cM). After construction of the best map, the colinearity and genetic distances of our genetic map were compared with published maps available at http://www.gramene.org. For comparative mapping, we used the genetic positions (cM) of SSR markers from Gramene (http://www.gramene.org). The syntenic regions were plotted using RCircos package (Zhang et al. 2013).
Recombination frequency
Recombination analysis was performed by R/qtl program (Broman et al. 2003) to investigate its relationship with genetic distance, segregation distortion, and linkage disequilibrium. The recombination fraction between each pair of markers (\(\hat{\rm{r}}\)) was estimated, and a LOD score for the test of \(\hat{\rm{r}}\) = 1/2 for markers was calculated. The pairwise recombination fractions were plotted versus LOD scores using the ‘plot.rf’ function. Markers on various chromosomes should remain unlinked, and for markers on the same chromosome, it was expected to obtain smaller recombination fraction for more closely linked markers.
Linkage disequilibrium
We tested the evidence of linkage disequilibrium among the markers on the same chromosome using the GGT 2.0 program (Berloo 2008). Linkage disequilibrium between pairs of loci was estimated by the D′ measure, its application in earlier studies, and the r2 measure, its usage by population geneticists (Dawson et al. 2002). Furthermore, statistical models were adapted to identify the map distance across which linkage disequilibrium can occur within our mapping population. RCircos package was also used to create a graphical view of the extent and pattern of segregation distortion, linkage disequilibrium (r2 and D′), and recombination frequency across three chromosomes assembled in this study (Zhang et al. 2013).
Results
Marker analysis and pattern of segregation distortion
To examine the chromosomal regions carrying the known scent QTLs, we selected microsatellite and flanking markers reported for such QTLs in order to not only validate but also explore specific loci for this appealing trait. However, most of the screened markers were monomorphic. As out of 112 SSR markers that covered chromosomes 3, 4 and 8 of rice molecular linkage groups, 23 (20.53 %) markers (6, 6 and 11 markers located on chromosomes 3, 4, and 8, respectively) indicated polymorphism between parental lines. However, because these polymorphic markers were properly scattered in the respective genomic regions related to aroma, we may infer our important findings.
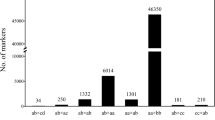
Of the polymorphic loci, eight markers exhibited significant segregation distortion (P < 0.01). Based on the cluster analysis, Fig. 1 indicates the 23 loci were clustered into three particular groups according to their direction towards genotypes. These groups were marked as Cluster I (B), Cluster II (H), and Cluster III (A). Four (17.4 %) markers deviated towards MR84 (A), and three (13 %) markers towards MRQ74 (B). The largest cluster, Cluster II, comprising 69.6 % of the loci, displayed a predominance of heterozygotes.

Clustered markers based on their preferences towards alleles
Studies on the marker loci segregation patterns revealed that the distorted segregations were not randomized (P < 0.01). For every locus, the identified segregations were investigated against the expected Mendelian ratios for the frequency of alleles (p = q) and the distribution of genotype frequencies (p2:2pq:q2) in order for examining whether there existed gametic or zygotic selections. 15 (65.2 %) mapped loci fitted the expected Mendelian segregation, while 8 (34.8 %) deviated from it (P < 0.01). All markers (100 %) on chromosome 8 displayed the expected predominance of H genotypes. The allele frequencies of these eight markers significantly distorted, but their F2 genotype frequencies did not show any significant distortion. Thus, we concluded that selective transmission of either female or male gamete likely leads to segregation distortion in this region.
The results indicated the distribution of the distorted markers mainly located on the third and fourth chromosomes, with 6 and 2 loci, respectively. Although the markers mapped on chromosomes 3 and 4 in the genetic map (100 and 33.3 %, respectively) indicated significant segregation distortion in the population, the most skewed segregation markers appeared on chromosome 3. All of the mapped markers on chromosome 3 were significantly deviated, with three showing an excess of AA genotypes, two locus having BB genotypes, and one loci with an excess of heterozygote. Most of the markers deviated towards the cultivated variety MR84 (A). Additionally, chromosome 4 had 16.7 % frequency of each genotype.
Linkage map construction and analysis of maps colinearity
Markers within the chromosomal area that surrounds the segregation distortion locus will always be affected by segregation distortion (Alheit et al. 2011). In other words, the non-Mendelian segregation of markers affects not only the estimation of genetic distance between two markers but also the order of markers on a same linkage group (Zhu et al. 2007). Hence, we aimed to compare genome structure of the chromosomes studied in this research with those from Gramene in order to reveal not only unique molecular genetics of species but also the possible effect of segregation distortion on genetic linkage map construction. The component maps were constructed from 23 SSR markers on the three rice chromosomes that were anticipated to possess aroma QTLs. The resulting linkage groups showed the total lengths of 290, 213.2, and 148.5 cM, however, 231, 152, and 124 cM were the overall lengths of Gramene map for chromosomes 3, 4, and 8, respectively. Genetic intervals between marker pairs from our study on chromosomes 3 (34.8, 30.9, 64.7, 116.3, 43.3) and 4 (45.3, 36.7, 59. 7, 43.7, 27.8) were higher than those intervals on chromosome 3 (11.7, 20.8, 32.8, 115.8, 14.6) and 4 (78.1, 8.3, 8.2, 1.4, 35.5) from Gramene, however, differences on chromosome 8 (23.8, 31.8, 10.4, 4, 1.3, 1.5, 7.2, 12.3, 28.7, 27.8) were moderately more than Gramene chromosome 8 (6.4, 5.7, 3, 8.1, 6.5, 5, 9.8, 5.9, 7.5, 20.9). Colinearity, as the collocation of marker in the same chromosome, was widely conserved along with each pair of same linkage group (Fig. 2), with the exception of few cases of inverted order (RM223 and RM515 on chromosome 8) with small interval. However, the genetic distances between markers appeared to be unequal. While average and maximum genetic map distances between loci from Gramene map ranged from 39.1 to 115.8 cM (chromosome 3), 26.3 to 78.1 cM (chromosome 4), and 7.9 to 21 cM (chromosome 8), the genetic map generated from this study revealed average and maximum spacing from 58 to 116.3 (chr. 3), 42.6 to 59.6 (chr. 4), and 14.9 to 31.5 cM (chr. 8).

Conservation of synteny and linear order of markers in the two genetic maps. LG constructed linkage groups in the present study, HG previously published linkage groups (http://www.gramene.org)
Detection of linkage disequilibrium across three chromosomes
Linkage disequilibrium was calculated for the loci mapped onto the linkage map (Fig. 3). Most of the adjacent marker loci indicated high r2 and D′ values. When examining marker loci mapped onto various chromosomes, linkage disequilibrium indicating r2 and D′ > 0.5 was located on a region with 26.3 and 36.7 cM length on chromosome 8 including 20–21 locus pairs, respectively, whereas, the rest of loci pairs on three chromosomes were found with the modest linkage disequilibrium or uncorrelated across the population and hence were in linkage equilibrium (LE). Of the regions indicating significant linkage disequilibrium across chromosome 8, four (with r2 > 0.8) and seven (with D′ > 0.8) loci had extremely high linkage disequilibrium in comparison with other loci. The highest common values (>0.8) of both r2 and D′ measures showing 0.88, 0.90 (RM404-RM223), 0.83, 0.89 (RM515-RM223), 0.81, 0.87 (RM137-RM223), and 0.80, 0.86 (RM223-RM210) demonstrated the lowest recombination ratio 0.03, 0.04, 0.05, and 0.5, respectively.

A graphical visualization of the extent and pattern of segregation distortion, linkage disequilibrium (r2 and D′), and recombination frequency across constructed linkage groups. From outside to inside, the first, second, third, and fourth circles represent segregation distortion percentage, r2, Lewontin D′ values, and RF, respectively
Figure 4 revealed the observed relationship of the two linkage disequilibrium measures (r2 and D′) with distance for a population size of 189 F2s and codominant markers. Extensive linkage disequilibrium between markers appeared at short distances but it was reduced rapidly along with distance. This study also illustrated a correlation between extent of linkage disequilibrium and estimated recombination rate indicated in Fig. 5. The upper left and the lower right showed the estimated recombination fraction (\(\hat{\rm{r}}\)) between markers and the logarithm of odds scores (LOD), respectively. Red shows linked pairs of markers (low \(\hat{\rm{r}}\) or high LOD), and blue shows unlinked pairs (high \(\hat{\rm{r}}\) or low LOD). Some red points can be observed in the lower right, showing markers on chromosome 8 that appeared linked confirming those results of linkage disequilibrium.

The relationship of r2 versus distance was measured in the three targeted genomic regions. The plots on the left and right correspond to r2 and D′ values, respectively

Estimated pairwise recombination fractions (upper left) and LOD scores (lower right) for all pairs of markers on LGs 3, 4, and 8
Discussion
The original objective of the present research was to explore pattern and extent of segregation distortion and linkage disequilibrium across the known aroma loci. We could achieve this by developing an intercross mapping population and SSR markers. Then, a linkage map was established for analysis of molecular genetics in rice. Segregation distortion can be rooted in genetic and environmental factors, and the relative contribution of each of these factors can be varied in different populations (Xu et al. 1997). Moderate departures from frequencies 1:2:1 are typical and even can point out the presence of partially lethal alleles. Segregation distortion analysis in this research resulted in clusters of distorted markers on chromosomes 3 and 4. The eight distorted markers distributed on these two chromosome areas showed at least two segregating distortion loci in MR84/MRQ74. However, consistent with Paillard et al. (2003), a region with at least three adjacent loci indicating significant segregation distortion (P < 0.05) was identified as a segregation distortion region (SDR). Therefore, only one SDR, including six deviated loci, was identified in the third chromosome of rice. Significant segregation distortion in both F2 genotype frequency and allele frequency indicates both gametic and zygotic selections influence segregations. If only F2 genotype frequencies indicate significantly distortion but allele frequencies of markers do not show distortion, only zygotic selection affects the segregation. In contrast, if only allele frequencies indicate significantly distortion but their F2 genotype frequencies do not indicate any significant distortion, the gametic selection has great influence on the segregations. In our research, there were eight markers whose F2 genotype frequencies did not show any significant distortion but their allele frequencies were significantly distorted. Thus, we concluded that selective transmission of either female or male gamete likely leads to segregation distortion in this region. Consistent with our finding, Harushima et al. (1996) mentioned that zygote eliminator gene is not engaged in segregation distortion on this area. Because the distorted markers were mapped near to ga or hybrid sterility (S) genes, distortion might be due to ga or S genes as reported in several studies (Matsushita et al. 2003; Reflinur et al. 2014; Shanmugavadivel et al. 2013; Wang et al. 2009; Wu et al. 2010; Xu et al. 1997). Since F2 population contains heterozygotes, and heterozygote and dominant homozygote are not easily distinguishable, mapping accuracy decreases (Zhao et al. 2006). Although segregation distortion is a very complicated phenomenon, the effect of ga or S is yet observed as its main cause in this population. However, in order to have a better understanding of segregation distortion factors, further investigations on different populations and places are suggested for future studies.
Nakagahra (1972) firstly observed segregation distortion on chromosome 3 and reported that Gametophytic genes (ga2 and ga3), which were distinguished by the allele frequency (P1 = P2) in F2 populations, were the causes of this distortion. Hitherto, 15 gametophyte genes (ga1 ~ ga14 and gax) have been mapped on different chromosomes (Wu et al. 2010). Ga2, ga3, and ga14 were mapped on the third chromosome whereas ga6, ga10, and ga12 were located on the fourth chromosome of rice. Among these segregation distorter genes, the ones that led to segregation distortion in this population could be inferred by comparative analysis and physical interval of tightly linked markers (http://www.gramene.org). However, it seems to be complicated to conclude how many reproductive obstacles exist in the population. Most of the markers associated with the gametophytic and hybrid sterility obstacles indicated favoring homozyotes of the female parent (A) in this population. The most deviated markers, RM85 and RM514, on the end long arm of chromosome 3 may have corresponded to ga14. Harushima et al. (1996) and Nakagahra (1972) reported ga2 in chromosome 3, at the interval of 9.9–15.17 Mb. Some of the reported SDRs were placed within the SDR identified in this research using the same microsatellite markers, but on different populations developed from different genetic backgrounds in rice, and were thus validated. For instance, in our population, one of the most severe distorted loci (RM282) was on the short arm of chromosome 3 in the physical interval of ga2. Therefore, this distortion was concluded to be caused by ga2. In fact, the marker locus is as same as the marker locus previously found as the ga2 gene (Matsushita et al. 2003). Furthermore, whereas several deviation factors and chromosomal areas indicting segregation distortion have been determined in rice, no distortion factors other than ga2 and ga3 were mapped on the short arm of chromosome 3 (Xu et al. 1997). Additionally, RM218 and RM545 on chromosome 3 were previously identified as indicating segregation distortion in an F2 population derived from the reciprocal XMG and N34 cultivars (Wang et al. 2009). Likewise, RM280 on chromosome 4 was identified in a DH population (Caiapo × IRGC103544) (Aluko et al. 2004) and a recombinant inbred mapping population (Basmati 370 × IRBB60) (Shanmugavadivel et al. 2013).
Due to the relationship between segregation distortion and aroma inheritance, there is a variety of different reports on the inheritance of fragrance. One of the most important factors causing such contradictory findings is segregation distortion (Lorieux et al. 1996; Sakthivel et al. 2009). The segregation pattern of a gene can be modified by segregation distortion. Also, it is possible that the two-gene segregations reported by Pinson (1994) were because of this phenomenon (Lorieux et al. 1996). In this research, the deviation in favor of non aromatic lines was also due to strong segregation distortion. Besides the inheritance of aroma, the power of QTL detection is also affected by segregation distortion; however, it does not produce false positive detection. Apart from these important issues, segregation distortion affects both the map length of chromosomes and markers order (Lorieux et al. 1996). Because of such critical reasons, we also performed comparative mapping to see the possible effect of segregation distortion on our genetic map. We compared genome structure of the chromosomes studied in this research with those from Gramene. This comparative map indicated the phenomena of molecular genetics of species with various genome structures. Predominant synteny was observed, and 21 markers were mapped in a common manner between the two studies, except a translocation between two highly adjacent loci (1.5 cM) located on chromosome 8 (Fig. 2). This indicated that segregation distortion did not disturb the ordering calculations. The observed genetic distances seemed to be higher than those from Gramene map, which in turn, increased lengths of the selected linkage groups were observed. Although chromosome 8 represented a moderate synteny, as, some genetic intervals were found to be lower than Gramene map, differences on chromosomes 3 and 4 were higher than those intervals from Gramene. Irrespective of the fact that the linkage distance between markers differentiates at some chromosomal regions in different crosses and populations, but the order of markers is highly conserved (Antonio et al. 1996; Reflinur et al. 2014). However, it is likely that the increased lengths of chromosomes 3 and 4 are due to segregation distortion indicating segregation distortion affects the map length of chromosomes (Fig. 2). Hence, with knowledge regarding segregation distortion in a segregating population, we can control and manage the possible destructive impacts of segregation distortion on not only the fragrance inheritance (the segregation pattern of aroma genes) but also power of aroma QTLs detection, the estimation of recombination fractions, the map length, and markers order.
Apart from segregation distortion, we also discovered the presence of linkage disequilibrium across the respective loci on the map. Based on the results of this study, linkage disequilibrium was observed only in the chromosome 8. Interestingly, markers including very strong linkage were localized in the high marker density region of chromosome 8, and with the lowest genetic intervals. A chromosomal area with 26.3 and 36.7 cM length located on chromosome 8 indicated linkage disequilibrium with r2 and D′ > 0.5, while, the modest linkage disequilibrium or uncorrelated was observed in other loci pairs on three chromosomes. Although no large-scale assessment of linkage disequilibrium has been observed in O. sativa, the seminal research indicated a linkage disequilibrium decay of 100 kb around a disease resistance locus in the aus subpopulation (Garris et al. 2003), more recently of 50 kb in indica, of 5 kb in O. Rufipogon (Rakshit et al. 2007), 2 Mb in indica and tropical japonica, and 500 kb for O. Rufipogon (Caicedo et al. 2007). The physical extent of linkage disequilibrium around a gene defines the efficiency of linkage disequilibrium mapping, and it is the consequence of several factors, including the degree of artificial or natural selection on the gene or region of the genome, the rate of outcrossing, recombination fraction, the age of the allele under study, chromosomal location, and population size and structure (Garris et al. 2003). Across chromosome 8, four and seven markers had r2 and D′ more than 0.8, respectively, showing extremely high linkage disequilibrium. Moreover, comparative analysis was performed based on physical mapping of these markers to fragrance QTL/gene (fgr) on chromosome 8, according to previous reports. In this study, the chromosomal regions of markers with high linkage disequilibrium were compared to the aroma QTL reported in earlier studies on rice. Some of these genomic regions (RM223 and RM515) were mostly related to chromosomal region underlying fragrance QTL. Consequently, marker loci showing high linkage disequilibrium at short intervals can maintain high linkage disequilibrium with linked QTLs (also consistent with the results of our preliminary aroma QTL mapping) and thus are highly applicable for MAS. The significant QTLs influencing fragrant grain quality can be highly utilized by breeders for crop improvement programs and the further fine mapping of particular genes to enhance gene-based perfect markers for use in rice breeding and the mining of better alleles for these genes in aromatic rice collections.
Based on previous studies, the closer linked markers, the smaller the estimated recombination fraction (Broman and Sen 2009). We also observed high linkage disequilibrium between marker pairs at short distances (Fig. 4), as extreme linkage disequilibrium between loci appeared at the shortest distances, and decreased rapidly with distance. Similar descents were observed when using D′. The amount to which loci are linked can be measured using linkage disequilibrium. Linkage disequilibrium (due to linkage) is the net result of all the recombination events (Gupta et al. 2005), in contrast, linkage between loci can be broken up by recombination. Thus, the linkage disequilibrium level between the two loci will remain constant over time depending on the genetic distance, the recombination rate, and the effective population size (Soto-Cerda and Cloutier 2012). Regions with extensive linkage disequilibrium coincide strongly with recombination-suppressed regions and, conversely, regions with low linkage disequilibrium coincide with recombination-intense regions (Ostberg et al. 2013). Similarly, from our research, the highest linkage disequilibrium values, showing more than 0.8 r2 and D′ measures, indicated the lowest recombination ratio ranging from 0.03 to 0.5. Nevertheless, it sometimes happens that the rate of recombination between nearby loci increases. As there are a number of markers that are strongly associated with other markers, but with recombination fractions well above 1/2 (Broman and Sen 2009). This is the result of either gene conversion (Padhukasahasram et al. 2004; Gupta et al. 2005) or markers with switched alleles (A ↔ B) (Broman and Sen 2009). The high rate of gene conversion will often lead to breakdown of allelic associations across short distances, thus reducing the magnitude of linkage disequilibrium between closely linked adjacent loci (Gupta et al. 2005). A plot of the LOD scores against the estimated recombination fractions for all marker pairs not only showed a negative correlation between linkage disequilibrium and recombination but also gave a good view of possibility of this problem (Fig. 5).
Interestingly, besides gene conversion and switched alleles, the association of segregation distortion skewed to the same or different genotypes at the consecutive markers is another important reason for the observed recombination fraction being higher or lower than the intrinsic frequency. Segregation distortion influences the frequency of recombination between each pair of markers (Wu et al. 2010), as it has a remarkable effect on the estimation of recombination fractions on chromosomes which are carrying scent genes. As Lorieux et al. (1996) used the corrected estimation of recombination frequency on the 8th chromosome of rice in their analysis. Our results also indicated the presence of segregation distortion led to high recombination events. In the presence of appreciable segregation distortion, unlinked markers can appear to be linked (Broman and Sen 2009). As indicated in Fig. 3, regions which displayed high segregation distortion had high recombination frequency.
Simultaneously, the results of our analyses indicated that the presence of segregation distortion led to linkage equilibrium, proposing that distortion influences linkage between adjacent markers. A strong negative correlation was also observed between linkage disequilibrium and recombination frequency, as extreme linkage disequilibrium appeared at suppressed recombination at the shortest distances across genomic regions (Fig. 3). On the other hand, regions with high segregation distortion had low linkage disequilibrium and vice versa. Consequently, based on the findings of this study, segregation distortion directly influences recombination frequency, and recombination has also direct effect on linkage disequilibrium. Thus, it can be inferred that segregation distortion has indirectly a remarkable effect on linkage disequilibrium. Hence, there is an indirect relationship between segregation distortion and linkage disequilibrium. Surprisingly, broad-scale recombination and subsequently narrow-scale linkage disequilibrium could indicate the presence of segregation distortion. Additionally, we concluded that linkage disequilibrium can provide an estimate of the segregation distortion ratio of the genotypes of offspring (Fig. 3). Hence, linkage disequilibrium maps are useful for the identification of genomic regions that have undergone differential selection and related phenomena (Collins 2007).
Although segregation distortion is one of the important factors having remarkable effects on the controversial issues of fragrance trait (Lorieux et al. 1996; Sakthivel et al. 2009), linkage disequilibrium also plays an important role in clarification of these barriers. Previous studies have reported the presence of segregation distortion and linkage disequilibrium on the whole genome of rice, but nobody has reported these aspects across the known fragrance chromosomal regions. The novelty of the present study is in investigating the presence, extent, and relationship between segregation distortion and linkage disequilibrium across the genomic regions that contain fragrance QTLs. High linkage disequilibrium correlations between markers at short distances might indicate that such markers are able to hold high linkage disequilibrium with linked QTLs and are effective for MAS and QTL mapping analysis. Additionally, we discovered an indirect relationship between segregation distortion and linkage disequilibrium, and introduced linkage disequilibrium maps as useful estimator tools for segregation distortion. However, because there is no previous research and findings regarding the relationship between marker distortion and linkage disequilibrium, our finding is a key basic step towards further understanding and research on this important and useful issue in future. The knowledge generated by this study offers valuable resources for genetic mapping and molecular breeding for rice fragrant trait in future studies. Nevertheless, in order to precisely identify the allelic series of segregation distortion genes on chromosome 3, fine mapping of such genes is the next stage toward their cloning. Additionally, further QTLs analysis of gametophyte genes can develop our knowledge in the process of segregation distortion, and permit breeders to forecast the proper population size, and finally get the desired recombinants. Future research of linkage disequilibrium in crop plants will also clarify the structures of plant genomes and promote the use of MAS and map based cloning of genes for various features.
References
Alheit KV, Reif JC, Maurer HP, Hahn V, Weissmann EA, Miedaner T, Würschum T (2011) Detection of segregation distortion loci in triticale (x Triticosecale Wittmack) based on a high-density DArT marker consensus genetic linkage map. BMC Genomics 12:380
Aluko G, Martinez C, Tohme J, Castano C, Bergman C, Oard JH (2004) QTL mapping of grain quality traits from the interspecific cross Oryza sativa × O. glaberrima. Theor Appl Genet 109:630–639
Amarawathi Y, Singh R, Singh AK, Singh VP, Mohapatra T, Sharma TR, Singh NK (2008) Mapping of quantitative trait loci for basmati quality traits in rice (Oryza sativa L.). Mol Breed 21:49–65
Antonio BA, Inoue T, Kajiya H, Nagamura Y, Kurata N, Minobe Y et al (1996) Comparison of genetic distance and order of DNA markers in five populations of rice. Genome 39:946–956
Baumbach J, Rogers JP, Slattery RA et al (2012) Segregation distortion in a region containing a male-sterility, female-sterility locus in soybean. Plant Sci 195:151–156
Berloo RV (2008) GGT 2.0: versatile software for visualization and analysis of genetic data. J Hered 99:232–236
Bradbury LM (2009) Identification of the gene responsible for fragrance in rice and characterisation of the enzyme transcribed from this gene and its homologs. Dissertation, Southern Cross University
Bradbury LM, Henry RJ, Jin Q, Reinke RF, Waters DL (2005) A perfect marker for fragrance genotyping in rice. Mol Breed 16:279–283
Broman KW, Sen S (2009) A guide to QTL mapping with R/qtl. Springer, New York
Broman KW, Wu H, Sen Ś, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890
Brummer EC, Bouton JH, Kochert G (1993) Development of an RFLP map in diploid alfalfa. Theor Appl Genet 86:329–332
Caicedo AL, Williamson SH, Hernandez RD et al (2007) Genome-wide patterns of nucleotide polymorphism in domesticated rice. PLoS Genet 3:e163
Carlborg Ö, Jacobsson L, Åhgren P, Siegel P, Andersson L (2006) Epistasis and the release of genetic variation during long-term selection. Nat Genet 38:418–420
Collins AR (2007) Linkage disequilibrium and association mapping: an introduction. In: Collins AR (ed) Linkage disequilibrium and association mapping. Methods in molecular biology, vol 376. Humana Press, Totowa, pp 1–15
Dawson E, Abecasis GR, Bumpstead S et al (2002) A first-generation linkage disequilibrium map of human chromosome 22. Nature 418:544–548
Feuillet C, Keller B (2002) Comparative genomics in the grass family: molecular characterization of grass genome structure and evolution. Ann Bot 89:3–10
Garris AJ, McCouch SR, Kresovich S (2003) Population structure and its effect on haplotype diversity and linkage disequilibrium surrounding the xa5 locus of rice (Oryza sativa L.). Genetics 165:759–769
Gawel N, Jarret R (1991) A modified CTAB DNA extraction procedure for Musa and Ipomoea. Plant Mol Biol Rep 9:262–266
Golestan Hashemi FS, Rafii MY, Ismail MR et al (2013) Biochemical, genetic and molecular advances of fragrance characteristics in rice. Crit Rev Plant Sci 32:445–457
Gupta PK, Rustgi S, Kulwal PL (2005) Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol Biol 57:461–485
Harushima Y, Kurata N, Yano M, Nagamura Y, Sasaki T, Minobe Y, Nakagahra M (1996) Detection of segregation distortions in an indica-japonica rice cross using a high-resolution molecular map. Theor Appl Genet 92:145–150
Knox M, Ellis T (2002) Excess heterozygosity contributes to genetic map expansion in pea recombinant inbred populations. Genetics 162:861–873
Kumar S, Gill BS, Faris JD (2007) Identification and characterization of segregation distortion loci along chromosome 5B in tetraploid wheat. Mol Genet Genomics 278:187–196
Lorieux M, Petrov M, Huang N, Guiderdoni E, Ghesquière A (1996) Aroma in rice: genetic analysis of a quantitative trait. Theor Appl Genet 93:1145–1151
Lu H, Romero-Severson J, Bernardo R (2002) Chromosomal regions associated with segregation distortion in maize. Theor Appl Genet 105:622–628
Lyttle T (1991) Segregation distorters. Annu Rev Genet 25:511–557
Matsushita S, Iseki T, Fukuta Y, Araki E, Kobayashi S, Osaki M, Yamagishi M (2003) Characterization of segregation distortion on chromosome 3 induced in wide hybridization between indica and japonica type rice varieties. Euphytica 134:27–32
McCouch SR, Teytelman L, Xu Y et al (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res 9:199–207
Nakagahra M (1972) Genetic mechanism on the distorted segregation of marker genes belonging to the eleventh linkage group in cultivated rice. Jap J Breed 22:232–238
Ollitrault P, Terol J, Chen C et al (2012) A reference genetic map of C. clementina hort. ex Tan.; citrus evolution inferences from comparative mapping. BMC Genomics 13:593
Ostberg CO, Hauser L, Pritchard VL, Garza JC, Naish KA (2013) Chromosome rearrangements, recombination suppression, and limited segregation distortion in hybrids between Yellowstone cutthroat trout (Oncorhynchus clarkii bouvieri) and rainbow trout (O. mykiss). BMC Genomics 14:570
Padhukasahasram B, Marjoram P, Nordborg M (2004) Estimating the rate of gene conversion on human chromosome 21. Am J Hum Genet 75:386–397
Paillard S, Schnurbusch T, Winzeler M et al (2003) An integrative genetic linkage map of winter wheat (Triticum aestivum L.). Theor Appl Genet 107:1235–1242
Paterson AH, Lander ES, Hewitt JD, Peterson S, Lincoln SE, Tanksley SD (1988) Resolution of quantitative traits into Mendelian factors by using a complete linkage map of restriction fragment length polymorphisms. Nature 335:721–726
Pinson S (1994) Inheritance of aroma in six rice cultivars. Crop Sci 34:1151–1157
Rakshit S, Rakshit A, Matsumura H et al (2007) Large-scale DNA polymorphism study of Oryza sativa and O. rufipogon reveals the origin and divergence of Asian rice. Theor Appl Genet 114:731–743
Reflinur Kim B, Jang SM et al (2014) Analysis of segregation distortion and its relationship to hybrid barriers in rice. Rice 7:3
Sakthivel K, Sundaram R, Shobha Rani N, Balachandran S, Neeraja C (2009) Genetic and molecular basis of fragrance in rice. Biotechnol Adv 27:468–473
Shanmugavadivel PS, Mithra SVA, Dokku P et al (2013) Mapping quantitative trait loci (QTL) for grain size in rice using a RIL population from Basmati × indica cross showing high segregation distortion. Euphytica 194:401–416
Soto-Cerda BJ, Cloutier S (2012) Association mapping in plant genomes. In: Caliskan M (ed) Genetic diversity in Plants. InTech, Rijeka, pp 29–54
Sun S, Gao FY, Lu XJ, Wu XJ, Wang XD, Ren GJ, Luo H (2008) Genetic analysis and gene fine mapping of aroma in rice (Oryza sativa L. Cyperales, Poaceae). Genet Mol Biol 31:532–538
R Core Team (2011) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. http://cranr-project.org
Temnykh S, Park WD, Ayres N et al (2000) Mapping and genome organization of microsatellite sequences in rice (Oryza sativa L.). Theor Appl Genet 100:697–712
Wang S, Tan Y, Tan X, Zhang Z, Wen J, Kou S (2009) Segregation distortion detected in six rice F2 populations generated from reciprocal hybrids at three altitudes. Genet Res 91:345–353
Wu YP, Ko PY, Lee WC et al (2010) Comparative analyses of linkage maps and segregation distortion of two F2 populations derived from japonica crossed with indica rice. Hereditas 147:225–236
Xu S (2008) Quantitative trait locus mapping can benefit from segregation distortion. Genetics 180:2201–2208
Xu S, Hu Z (2009) Mapping quantitative trait loci using distorted markers. Int J Plant Genomics. doi:10.1155/2009/410825
Xu Y, Zhu L, Xiao J, Huang N, McCouch SR (1997) Chromosomal regions associated with segregation distortion of molecular markers in F2, backcross, doubled haploid, and recombinant inbred populations in rice (Oryza sativa L.). Mol Gen Genet 253:535–545
Yi M, Nwe KT, Vanavichit A, Chai-arree W, Toojinda T (2009) Marker assisted backcross breeding to improve cooking quality traits in Myanmar rice cultivar Manawthukha. Field Crops Res 113:178–186
Zhang H, Meltzer P, Davis S (2013) RCircos: an R package for Circos 2D track plots. BMC Bioinform 14:1–5
Zhao B, Deng QM, Zhang QJ, Li JQ, Ye SP, Liang YS, Peng Y, Li P (2006) Analysis of segregation distortion of molecular markers in F2 population of rice. Acta Genet Sin 33:449–457
Zhao H, Nettleton D, Dekkers J (2007) Evaluation of linkage disequilibrium measures between multi-allelic markers as predictors of linkage disequilibrium between single nucleotide polymorphisms. Genet Res 89:1–6
Zhu C, Wang C, Zhang YM (2007) Modeling segregation distortion for viability selection I. Reconstruction of linkage maps with distorted markers. Theor Appl Genet 114:295–305
Acknowledgments
The authors express their acknowledgements of the Long-term Research Grant Scheme (LRGS), Food Security Project, Ministry of Higher Education, Malaysia, for the financial support to conduct activities on rice research program.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Golestan Hashemi, F.S., Rafii, M.Y., Ismail, M.R. et al. Comparative mapping and discovery of segregation distortion and linkage disequilibrium across the known fragrance chromosomal regions in a rice F2 population. Euphytica 204, 557–569 (2015). https://doi.org/10.1007/s10681-014-1322-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-014-1322-y