
Abstract
Bacterial canker is one of the most important diseases of stone fruit trees in various locations of Kurdistan province, Iran. Genetic diversity and evolutionary relationships among 20 fluorescent pseudomonads isolated from stone fruit trees with symptoms similar to bacterial canker were investigated using a polyphasic approach by means of phenotypic characterizations, repetitive PCR using the REP and ERIC primers and multilocus sequence typing (MLST) of four housekeeping genes (gapA, rpoD, gyrB and gltA). The pathogenicity of strains was carried out under greenhouse conditions. Twelve strains produced an expected amplified DNA fragment of about 752-bp which indicated the presence of the syrB gene. Based on MLST, these strains belonged to P. syringae species complex and included in the genomospecies 1, phylogroup 2b and 2d. Phylogenetic analysis of the other eight fluorescent pseudomonad strains by using gyrB and rpoD sequences allowed the identification of strains into P. fluorescens, P. putida and P. lutea groups. Unweighted pair group method analysis (UPGMA) of genomic fingerprints obtained by rep-PCR revealed 17 different patterns which grouped P. syringae strains into three clusters clearly separated from other fluorescent pseudomonads. MLST confirmed the genetic variability among strains obtained by rep-PCR. Grouping identified of P. syringae strains by both rep-PCR and MLST was related to geographic locations of strains.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Pseudomonas includes various species living as parasites or saprophytes in diverse terrestrial and aquatic habitats including plants (Marcelletti and Scortichini 2014). Partial sequence analysis of the housekeeping gene of Pseudomonas species allowed the discrimination of two lineages called P. aeroginosa and P. fluorescens. Both lineages were divided into different groups (Mulet et al. 2010). Some Pseudomonas spp. are responsible for the worldwide disease of many herbaceous and woody crop species. Amongst these, P. syringae as species complex cause more than a hundred diseases in unrelated plant species and disease incidence caused by this bacterium ranges from 50 to 100% in epidemic years (Lamichhane et al. 2015). This species complex is divided into different pathovars based on the plant hosts from which they were originally isolated (Young 2010). This type of classification has a practical usefulness but is more or less complicated. To clarify the taxonomic relationships within the pathovars, strains were divided to nine genomospecies based on DNA relatedness (Gardan et al. 1999). Several researches have explained other genetic techniques such as amplified fragment length polymorphism (AFLP), randomly amplified polymorphic DNA (RAPD) or specific primers corresponding to 16 s–23 s rDNA inter-transcribed spacer region (ITS) to identify bacterial isolates instead of DNA pairing studies (Clerc et al. 1998; Rees-George et al. 2010; Gutierrez-Barranquero et al. 2013). Repetitive sequence PCR (rep-PCR) analysis shows genetic diversity among P. syringae species complex (Louws et al. 1994; Vicente and Roberts 2007; Martin-Sanz et al. 2013). Multilocus sequence typing (MLST) is an alternative molecular typing method to characterize the phylogenetic relationship of bacteria at inter- and or intraspecies levels (Maiden 2006). MLST is based on comparing nucleotide sequences belonging to the core genome (Maiden et al. 1998). The first MLST analysis in respect to the seven housekeeping gene sequences, rpoD, gyrB, gltA, gapA, pgi, pfk and acn1 indicated that the species is highly clonal and stable (Sarkar and Guttman 2004). Other reports showed that MLST has the ability to assign P. syringae strains to their corresponding genomospecies and pathovars (Bull et al. 2011). MLST reveals a higher genetic variability than other methods such as rep-PCR (Ferrante and Scortichini 2010) and is a powerful tool for phylogenetic study of virulence, resistance and other ecological-associated phenotypes of P. syringae (Hwang et al. 2005). P. syringae is recognize as a complex of 13 phylogroups which differentiate according to the nucleotide sequences of one or more housekeeping genes (Berge et al. 2014).
Bacterial canker disease of stone fruit trees cause by P. syringae pvs. Syringae and morsprunorum and is an important disease worldwide. The syringae pathovar contains pathogenic strains to unrelated plant species including stone fruits, pome fruits, other woody plants and crop plants (Kennelly et al. 2007). Strains of this pathovar belong to genomospecies 1 (Gardan et al. 1999); additionally, MLST analysis indicates that strains clustered within phylogroup 2 (Hwang et al. 2005) and can be separated into four phylogenetic subgroups (Berge et al. 2014). Analysis of strains isolated from woody and herbaceous plants by using rep-PCR fingerprinting revealed a high level of genetic variability among strains (Scortichini et al. 2003). Strains of P. s. pv. syringae has been reported as the causal agent of disease in various locations of Iran (Shamsbakhsh and Rahimian 1997; Abbasi et al. 2013). Since 2005, a disease with similar symptoms to bacterial canker was observed on apricot (Prunus armeniaca) and peach (Prunus persica) trees in restricted areas of Kurdistan Province. A preliminary survey showed that P. syringae pv. syringae is the causal agent (Karimi-Kurdistani and Harighi 2008). Since then disease has become widespread in of the province and sever dieback and canker on branches were observed on stone fruit trees such as plum (P. domestica), nectarine (P. persica var. nectarina), almond (P. amygdalus), cherry (P. avium L.) and sour cherry (P. cerasus L.).
The objectives of this study were to characterize and identify fluorescent Pseudomonas strains isolated from infected stone fruit trees by phenotypic properties and multilocus sequence typing (MLST). The strains were further assessed for the presence of the syrB gene. Another objective was to assess the genetic diversity among strains using rep-PCR.
Materials and methods
Bacterial isolation and characterization
Bacteria were isolated from infected stem and twig tissues of stone fruit trees with symptoms similar to bacterial canker during April to July 2013 and 2014 in various locations in Kurdistan Province (Table 1). Infected tissues were surface sterilized in 0.5% sodium hypochlorite for 30 s, rinsed in sterile-distilled water, macerated in a small amount of distilled water and the suspension was spread onto KB medium (King et al. 1954). The plates were incubated at 26–28 °C for 3 days and fluorescent colonies were purified in the same medium. Bacteria were identified by biochemical and physiological tests including LOPAT (levan production, oxidase reaction, potato soft rot, arginine dihydrolase, tobacco hypersensitivity) and GATTa tests (gelatin hydrolysis, aesculin hydrolysis, tyrosinase activity, tartrate utilization) according to methods previously described (Schaad et al. 2001). Furthermore, the possible presence of the syrB gene was determined by PCR using the primers B1 and B2 (Sorensen et al. 1998).
Pathogenicity test
Bacterial strains grown in nutrient broth at 27 °C for 48 h were centrifuged (5 min, 7000 rpm) and the pellet was suspended in sterile-distilled water to a concentration of approximately 1 × 107 CFU ml−1. Bacterial suspensions of about 20 μl were injected into the 8- to 10-week-old cut stems of peach and apricot using a 22-gauge sterile needle. Sterile water was used as a control. Plant materials were kept at 26 to 27 °C in a mist chamber and disease development was assessed up to 3 weeks.
Genomic DNA preparation
Genomic DNA of bacterial isolates was extracted from 24-h grown cultured cells in LB medium. A 5 ml of suspension was treated by adding SDS/lysozyme followed by three times DNA extraction with phenol: chloroform:isoamyl alchohol (25:24:1) and once with chloroform: isoamyl alchohol (24:1). Finally, DNA was precipitated with 0.1 volume 3 M sodium acetate (pH: 4.8) and two volumes absolute ethanol overnight at -20 °C. The suspension was centrifuged at 14000 rpm for 5 min, washed with 70% ethanol, dried and re-suspended in 50 μL TE buffer.
Multilocus sequence typing (MLST)
Amplification and sequencing of four housekeeping genes, gapA, gltA, gyrB and rpoD were performed using procedures previously described (Sarker and Guttman 2004; Hwang et al. 2005). The PCR products sequencing was carried out using an ABI3730XL DNA sequencer (Applied Biosystems). Sequences for each of the four genes were compared with the nucleotide sequences of the same loci present in PAMDB using the blast program at the PAMDB website (Almeida et al. 2010). Nucleotide sequences were aligned using the ClustalW program (Ramu et al. 2003) available in BioEdit Sequence Alignment Editor 7.0.9.0 software (Hall 2011). Alignments were manually adjusted where necessary. Bayesian, neighbor-joining and maximum-likelihood phylogenetic analysis were performed on individual gene sequences as well as on the concatenated datasets by using PAUP version 4.0b10 (Swofford 2003). A phylogenetic tree was constructed with the concatenated sequences of the four loci using the Bayesian, neighbor-joining and maximum-parsimony methods (bootstrap analysis with 1000 replicates was conducted) with the same program.
Repetitive PCR genomic fingerprinting (rep-PCR)
Genomic fingerprints were determined for each strain as described previously (Versalovic et al. 1991) using primers corresponding to REP and ERIC primer sets with some modifications. Amplification was carried out in a total volume of 25 μl containing 12.5 μl of master mix (Fermentas, Lithunia), 1 μl of each primer (10 pmol μl−1), 1 μl (approximately 40 ng) of genomic DNA and 10.5 μl of sterile distilled water. PCR was performed in a Bio Rad MJ mini thermocycler (BioRad, Hercules, California, USA) with the following program: 1 cycle at 95 °C for 2 min, 35 cycles at 92 °C for 30 s, annealing at 40 °C (REP) and 50 °C (ERIC) for 1 min, extension at 65 °C for 8 min, followed by final extension at 65 °C for 8 min. The PCR amplification products were separated by gel electrophoresis on 1.5% agarose gel in 1 × TAE buffer at 80 V. Gels were stained with ethidium bromide and DNA visualized on a gel imaging system (UVIdoc, Cambridge, UK). The PCR amplifications were performed twice and the clearly resolved bands present in both amplification products were recorded. Differences in amplified fragment of each strain were assessed visually. Cluster analysis was performed on a similarity matrix produced using the Dice coefficient (Dice 1945) and subjected to the unweighted pair group method with arithmetic average (UPGMA) clustering algorithm using NTSYSpc software (Exeter Software, New York, NY, USA), version 2.02e.
Results
Isolation and characterization of bacterial strains
Results of phenotypic properties of isolates are summarized in Table 1. In total, 20 strains were isolated from infected tissues. The majority of isolates were positive for levan production and hypersensitive reaction on tobacco leaves, and all isolates were negative for oxidase, potato soft rot reaction and arginine dihydrolase (LOPAT 1A group). Moreover, the majority of isolates were able to hydrolyze aesculin and gelatin but did not produce tyrosinase, and utilization of D-tartrate was negative. When pathogenicity test was carried out on cut stems of peach and apricot, twelve strains caused similar symptoms to bacterial canker on at least one of these plants 15–21 days after inoculation (Fig. 1). PCR amplification of the syrB gene was performed for all strains. Twelve strains produced a DNA fragment of about 752-bp which indicated the presence of the syrB gene (Fig. 2).

The result of the pathogenicity test performed on peach stem cutting. a negative control inoculated with sterile-distilled water, b plant material inoculated with bacterial isolate. Results were recorded up to 21 days after inoculation

Results of the PCR using primers B1 and B2 were produced the expected DNA fragment of about 752-bp which indicated the presence of syrB gene
MLST analysis
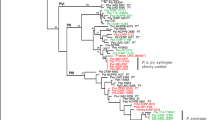
Polymerase chain reaction using primers corresponding to rpoD, gltA, gapA and gyrB genes generated amplified fragments of about 1118, 998, 650 and 748 bp, respectively. All sequences obtained in this study were deposited in the GenBank data library under the following accession numbers: gapA gene, KX160009 to 160,028; gltA, 160,029 to 160,048; gyrB, KX160049 to KX190068 and rpoD gene, KX160069 to KX160088. The concatenated sequences of four genes comprising of 2010 aligned nucleotides were analyzed by MLST. A phylogenetic tree was then built using the Bayesian, neighbor-joining and maximum parsimony algorithm (Fig. 3). MLST analysis of four housekeeping genes clearly indicated that 12 strains belong to P. syringae species complex and revealed genetic variability amongst these strains. All strains were grouped within a cluster which agreed with group II previously identified by Hwang et al. (2005) and genomospecies 1 of Gardan et al. (1999). According to MLST the P. syringae species complex strains separated into three subgroups (A, B and C). All strains in the same subgroup showed a genetic distance less of than 1% but genetic distance between subgroups was more than 2.5%. Subgroup A belonged to phylogroup 2b with the greatest similarity to PsyFF5 and subgroup B and C belonged to phylogroup 2d. Strains of subgroup B had the greatest similarity to syriHRIW 7924 whereas, subgroup C showed great similarity to syriHRIW 7872 and PsyB48 strains, previously identified (Clarke et al. 2010; Berge et al. 2014; Nowell et al. 2016). The grouping identified show direct relation to geographic areas of isolation. This analysis also clearly separated P. syringae species complex strains from other fluorescent pseudomonads isolated. Two housekeeping genes, gyrB and rpoD were selected for MLST analysis and phylogenetic relationship of eight fluorescent pseudomonad strains with selected type strains of Pseudomonas genus previously identified (Yamamoto et al. 2000; Mulet et al. 2010). Bayesian, neighbor-joining and maximum-parsimony analysis of the concatenated gyrB and rpoD sequences allowed the classification of isolates in different groups (Fig. 4). All isolates belonged to P. fluorescens lineage. Po76 clustered with P. orientalis, Pf95 clustered with P. fluorescent Pf5 and Pp42 showed the most similarity to P. putida type strains. Pg37 strain clustered with P. graminis type strain, and Pg53, Pg56, Pg81 and Pg82 belonged to the P. lutea group but failed to cluster with type strains within this group.

Phylogenetic tree of Pseudomonas syringae species complex and other Pseudomonas strains isolated from infected tissues based on the phylogenetic analysis of concatenated sequences gapA, rpoD, gyrB and gltA genes. Dendrogram was generated by Bayesian, neighbor-joining and maximum parsimony (BI/NJ/MP). Numbers above the nodes are bootstrap scores based on 1000 replicates. The five groups refer to those described by Berge et al. (2014) and Sarkar and Guttman (2004)

Phylogenetic tree of type strains of Pseudomonas species and Pseudomonas strains isolated from infected tissues based on the phylogenetic analysis of concatenated sequences rpoD and gyrB genes. Dendrogram was generated by Bayesian, neighbor-joining and maximum parsimony (BI/NJ/MP). Numbers above the nodes are bootstrap scores based on 1000 replicates. The groups refer to those described by Mulet et al. (2010)
REP and ERIC fingerprint analysis
REP- and ERIC-PCR generated 17 and 20 polymorphic bands ranging in size from 0.1 to 3 kb, respectively (Fig. 5a, b). The separate dendrograms obtained for REP, ERIC, and combined data had cophenetic correlation coefficients of 0.87, 0.89 and 0.91, respectively. UPGMA cluster analysis of the combined data obtained in the REP and ERIC-PCR experiments using Dice’s coefficient revealed that the strains with positive results for pathogenicity test and presence of syrB gene separated from other non-pathogenic strains at a similarity level of 45% and could also be clustered into three groups (I, II and III) at a similarity level of approximately 65% (Fig. 6). The grouping identified was related to the geographic locations of strains. Group I and III were formed by strains isolated from peach, nectarine, and plum in two regions (Sanandaj and Kamyaran). Group III included strains isolated from almond and plum trees of Marivan region.

a & b REP and ERIC-PCR fingerprints of fluorescent pseudomonads isolated from infected tissues. M: 1 kb DNA ladder

Dendrogram of genetic relatedness of the REP and ERIC fingerprint patterns generated by 20 strains studied. Cluster analysis was performed using Dice,s coefficient
Discussion
Identification, description and diversity of fluorescent pseudomonads isolated from infected stone fruit trees with symptoms similar to bacterial canker were assessed by phenotypic properties, detection of syrB gene and means of rep-PCR and MLST of four housekeeping genes, gapA, rpoD, gyrB and gltA. Our results show that P. s. pv. syringae is the main causal agent of disease in the areas studied. P. s. pv. syringae has the ability to infect a wide range of woody and herbaceous plant species (Kennelly et al. 2007). It suggests that this pathogen has a high degree of genetic variability. The syrB gene was detected in all the strains identified as P. s. pv. syringae. Although the presence of the syrB gene is a determinative characteristic to identify P. s. pv syringae, not all strains harbor this gene (Zeller et al. 1997; Sorensen et al. 1998).
Results of our study based on rep-PCR fingerprints showed genetic diversity among strains belonging to P. syringae species complex and clearly discriminated these strains from other fluorescent pseudomonads strains isolated from infected tissues. Although individual results of REP and ERIC-PCR fingerprints allow the separation of P. syringae species complex strains into different groups, combined dendrogram had higher cophenetic correlation coefficient showing that grouping is more accurate after combining the data. Previous studies also show that rep-PCR can differentiate P. syringae strains at the species and sub-species level (Louws et al. 1994; Weingart and Völksch 1997; Vicente and Roberts 2007). Despite the limited number of strains used in this study results illustrated that based on rep-PCR, P syringae species complex, strains were clustered into three groups and the grouping identified was related to geographic origin of the strains. Similarly, according to previous study, the rep-PCR technique differentiated phylotype of the pathovar syringae isolated from mango plants based on geographical origins (Gutierrez-Barranquero et al. 2013). Some other reports also show rep-PCR fingerprints diversity of P. syringae strains isolated from stone fruit trees and this diversity is in relation to host plants and place of isolation (Little et al. 1998; Scortichini et al. 2003; Kaluzna et al. 2010).
Multilocus sequence typing (MLST) based on sequencing of housekeeping genes is a powerful, universal and definitive method for characterization, diversity and pathogen population genetic study (Maiden 2006). MLST has previously been performed to compare the phylogeny of P. syringae species complex strains (Sarkar and Guttman 2004; Hwang et al. 2005; Gironde and Manceau 2012; Gutierrez-Barranquero et al. 2013; Berge et al. 2014; Marcelletti and Scortichini 2014). MLST of our data based on concatenated sequences of four genes revealed three subgroups amongst the P. syringae species complex strains. Although the number of strains was low, our results demonstrate that this strain is mainly related to geographical origin. This analysis also clearly confirmed the three groups identified by rep-PCR. All P. syringae species complex strains belonged to group II previously identified by Hwang et al. (2005). Our results showed that P. syringae species complex strains are divided into three subgroups (A, B and C). Strains from subgroup A are clustered with Psy FF5 isolated from ornamental pear supported with 96% value and belong to clade 2b of P. syringae. In addition, subgroups B and C strains are clustered with Psy B48 isolated from peach supported with 95% bootstrap value and belong to clade 2d (Fig. 2). Despite the subgrouping differentiation and differences in host plants, all strains had the same phenotypic properties. This finding is in agreement with the close relation of clade 2b and 2d both in genetic and phenotypic properties as previously reported (Berge et al. 2014).
It has been proposed that MLST based on housekeeping genes provide an accurate resolution of phylogenetic relationships among Pseudomonas species and facilitate the phylogenetic description of new isolates (Mulet et al. 2010; Tayeb et al. 2005). The aim of our study was also focused on differentiation of P. syringae species complex strains from other fluorescent Pseudomonads isolated from infected tissues based on MLST approach. Our results clearly indicate that analysis of the two concatenated genes (rpoD and gyrB) is sufficient for phylogenetic analysis of the species and differentiate fluorescent Pseudomonads from strains belonging to P. syringae species complex (Fig. 3). Representative strains of syringae, fluorescens, lutea and putida groups clustered separately from each other. Dendrogram built on with Bayesian, neighbor-joining and maximum parsimony methods provided the same tree topology and were supported by high bootstrap values. Previous reports also illustrate the considerable robustness of concatenated 16S rRNA, rpoD and gyrB genes for identification and sufficient phylogenetic analysis of Pseudomonas strains. However, in some cases the inclusion of rpoB gene might be necessary (Mulet et al. 2010; Gomila et al. 2015).
In conclusion, our analysis indicated that despite geographic origin or host plant, P. syringae species complex strains isolated from stone fruit trees are a clonal organism and MLST of housekeeping genes can be clearly discriminate these strains from other fluorescent Pseudomonads. Although some strains showed a close relationship to the already described species, some of the strains identified in the present study by MLST such as Pg53, Pg82, Pg56 and Pg81 represent phylogenetically independent clusters. To clarifying the exact position of these strains within the genus Pseudomonas it is necessary to describe the phenotypic properties and nucleotide sequences of other housekeeping genes. Until now, phylogenetic analysis of housekeeping genes is the most adapted method to identify strains belonging P. syringae species complex. However, several reports indicate that a simple and reliable PCR-based method allows for identification of P. syringae phylogroups (Borschinger et al. 2015) which might be useful for our case study in the future.
References
Abbasi, V., Rahimian, H., & Tajick-Ghanbari, M. A. (2013). Genetic variability of Iranian strains of Pseudomonas syringae pv. syringae causing bacterial canker disease of stone fruits. European Journal of Plant Pathology, 135, 225–235.
Almeida, N. F., Yan, S., Cai, R., Clarke, C. R., Morris, C. E., Schaad, N. W., Schuenzel, E. L., Lacy, G. H., Sun, X., Jones, J. B., Castillo, J. A., Bull, C. T., Leman, S., Guttman, D. S., João, C., Setubal, J. C., & Vinatzer, B. A. (2010). PAMDB, a multilocus sequence typing and analysis database and website for plant-associated microbes. Phytopathology, 100, 208–215.
Berge, O., Monteil, C. L., Bartoli, C., Chandeysson, C., Guilbaud, C., Sands, D. C., & Morris, C. E. (2014). A user's guide to a data base of the diversity of Pseudomonas syringae and its application to classifying strains in this phylogenetic complex. PloS One, 9, e105547.
Borschinger, B., Bartoli, C., Guilbaud, C., Parisi, L., Bourgeay, J. F., Buisson, E., & Morris, C. E. (2015). A set of PCRs for rapid identification and characterization of Pseudomonas syringae phylogroups. Journal of Applied Microbiology, 120, 714–723.
Bull, C. T., Clarke, C. R., Cai, R., Vinatzer, B. A., Jardini, T. M., & Koike, S. T. (2011). Multilocus sequence typing of Pseudomonas syringae sensu lato confirms previously described genomospecies and permits rapid identification of P. syringae pv. coriandricola and P. syringae pv. apii causing bacterial leaf spot on parsley. Phytopathology, 101, 847–858.
Clarke, C. R., Cai, R., Studholme, D. J., Guttman, D. S., & Vinatzer, B. A. (2010). Pseudomonas syringae strains naturally lacking the classical P. syringae hrp/hrc locus are common leaf colonizers equipped with an atypical type III secretion system. Molecular Plant Microbe Interaction, 23, 198–210.
Clerc, A., Manceau, C., & Nesme, X. (1998). Comparison of randomly amplified polymorphic DNA with amplified fragment length polymorphism to assess relatedness within genospecies III of Pseudomonas syringae. Applied and Environmental Microbiology, 64, 1180–1187.
Dice, L. R. (1945). Measures of the amount of ecologic association between species. Ecology, 26, 297–302.
Ferrante, P., & Scortichini, M. (2010). Molecular and phenotypic features of Pseudomonas syringae pv. actinidiae isolated during recent epidemics of bacterial canker on yellow kiwifruit (Actinidia Chinensis) in central Italy. Plant Pathology, 59, 954–962.
Gardan, L., Shafik, H., Belouin, S., Broch, R., Grimont, F., & Grimont, P. A. D. (1999). DNA relatedness among the pathovars of Pseudomonas syringae and description of Pseudomonas tremae sp. nov. and Pseudomonas Cannabina sp. nov. (ex Sutic and Dowson 1959). International Journal of Systematic Bacteriology, 49, 469–478.
Gironde, S., & Manceau, C. (2012). Housekeeping gene sequencing and multilocus variable number tandem-repeat analysis to identify subpopulations within Pseudomonas syringae pv. maculicola and Pseudomonas syringae pv. tomato that correlate with host specificity. Applied and Environmental Microbiology, 78, 3266–3279.
Gomila, M., Pena, A., Mulet, M., Lalucat, J., & Garcia-Valdes, E. (2015). Phylogenomics and systematics in Pseudomonas. Frontiers in Microbiology, 6, 214.
Gutierrez-Barranquero, J. A., Carrion, V. J., Murillo, J., Arrebola, E., Arnold, D. L., Cazorla, F. M., & de Vicente, A. A. (2013). Pseudomonas syringae diversity survey reveals a differentiated phylotype of the pathovar syringae associated with the mango host and mangotoxin production. Phytopathology, 103, 1115–1129.
Hall, T. (2011). BioEdit: an important software for molecular biology. GERF Bulletin of Biosciences, 2, 60–61.
Hwang, M. S., Morgan, R. L., Sarker, S. F., Wang, P. W., & Guttman, D. S. (2005). Phylogenetic characterization of virulence and resistance phenotypes of Pseudomonas syringae. Applied and Environmental Microbiology, 71, 5182–5191.
Kaluzna, M., Ferrante, P., Sobiczewski, P., & Scortichini, M. (2010). Characterization and genetic diversity of Pseudomonas syringae from stone fruits and hazelnut using repetitive-PCR and MLST. Journal of Plant Pathology, 92, 781–787.
Karimi-Kurdistani, G., & Harighi, B. (2008). Phenotypic and molecular properties of Pseudomonas syringae pv. syringae the causal agent of bacterial canker of stone fruit trees in Kurdistan province. Journal of Plant Pathology, 90, 81–86.
Kennelly, M. M., Cazorla, F. M., de Vicente, A., Ramos, C., & Sundin, G. W. (2007). Pseudomonas syringae diseases of fruit trees. Progress toward understanding and control. Plant Disease, 91, 4–17.
King, E. D., Ward, M. K., & Raney, D. E. (1954). Two simple media for the demonstration of pyocyanin and fluorescin. The Journal of Laboratory and Clinical Medicine, 44, 301–307.
Lamichhane, J. R., Messean, A., & Morris, C. E. (2015). Insights into epidemiology and control of diseases of annual plants caused by the Pseudomonas syringae species complex. Journal of General Plant Pathology, 81, 331–350.
Little, E. L., Bostock, R. M., & Kirkpatrick, B. C. (1998). Genetic characterization of Pseudomonas syringae pv. syringae strains from stone fruits in California. Applied and Environmental Microbiology, 64, 3818–3823.
Louws, F. G., Fulbright, D. W., Stephans, C. T., & Bruijn, F. G. (1994). Specific genomic fingerprinting of phytopathogenic Xanthomonas and Pseudomonas pathovar and strains generated with repetitive sequences and PCR. Applied and Enviromental Microbiology, 60, 2286–2295.
Maiden, M. C. (2006). Multilocus sequence typing of bacteria. Annual Review of Microbiology, 60, 561–588.
Maiden, M. C., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., Zhang, Q., Zhou, J., Zurth, K., Caugant, D. A., Feavers, I. M., Achtman, M., & Spratt, B. G. (1998). Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proceedings of the National Academy of Sciences of the USA, 95, 3140–3145.
Marcelletti, S., & Scortichini, M. (2014). Definition of plant-pathogenic Pseudomonas genomospecies of the Pseudomonas syringae complex through multiple comparative approaches. Phytopathology, 104, 1274–1282.
Martin-Sanz, A., de la Vega, M. P., Murillo, J., & Caminero, C. (2013). Strains of Pseudomonas syringae pv. syringae from pea are phyllogenetically and pathogenically diverse. Phytopathology, 103, 673–681.
Mulet, M., Lalucat, J., & Garcia-Valdes, E. (2010). DNA sequence-based analysis of the Pseudomonas species. Environmental Microbiology, 12, 1513–1530.
Nowell, R. W., Laue, B. E., Sharp, P. M., & Green, S. (2016). Comparative genomics reveals genes significantly associated with woody hosts in the plant pathogen Pseudomonas syringae. Molecular Plant Pathology, 17, 1409–1424.
Ramu, C., Sugawara, H., Koike, T., Lopez, R., Gibson, T. J., Higgins, D. G., & Thompson, J. D. (2003). Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Research, 13, 3497–3500.
Rees-George, J., Vanneste, J. L., Comish, D. A., Pushparajah, P. S., Yu, J., Templeton, M. D., & Everett, K. R. (2010). Detection of Pseudomonas syringae pv. actinidiae using polymerase chain reaction (PCR) primers based on the 16S-23S rDNA intertranscribed spacer region and comparison with PCR primers based on other gene regions. Plant Pathology, 59, 453–464.
Sarkar, S. F., & Guttman, D. S. (2004). Evolution of the core genome of Pseudomonas syringae, a highly clonal, endemic plant pathogen. Applied and Environmental Microbiology, 70, 1999–2012.
Schaad, N. W., Jones, J. B., & Chun, W. (2001). Laboratory guide for identification of plant pathogenic bacteria, third eds (p. 373). St Paul: APS Press.
Scortichini, M., Marchesi, U., Dettori, M. T., & Rossi, M. P. (2003). Genetic diversity, presence of the syrB gene, host preference and virulence of Pseudomonas syringae pv. syringae strains from woody and herbaceous host plants. Plant Pathology, 52, 277–286.
Shamsbakhsh, M., & Rahimian, H. (1997). Comparison of pathogenic strains of Pseudomonas syringae on various plants in Iran. Iranian Journal of Plant Pathology, 33, 133–143.
Sorensen, K. N., Kim, K. H., & Takemoto, J. Y. (1998). PCR detection of cyclic lipodepsinonapeptide-producing Pseudomonas syringae pv. syringae and similarity of strains. Applied and Environmental Microbiology, 64, 226–230.
Swofford, D. L. (2003). PAUP* 4.0b10: phylogenetic analysis using parsimony (*and other methods). Sunderland: Sinauer Associates.
Tayeb, L., Ageron, E., Grimont, F., & Grimont, P. A. D. (2005). Molecular phylogeny of the genus Pseudomonas based on rpoB sequences and application for the identification of isolates. Research in Microbiology, 156, 763–773.
Versalovic, J., Koeuth, T., & Lupski, J. R. (1991). Distribution of repetitive DNA-sequences in eubacteria and application to fingerprinting of bacterial genomes. Nucleic Acids Research, 19, 6823–6831.
Vicente, J. G., & Roberts, S. J. (2007). Discrimination of Pseudomonas syringae isolates from sweet and wild cherry using rep-PCR. European Journal of Plant Pathology, 117, 383–392.
Weingart, H., & Völksch, B. (1997). Genetic fingerprinting of Pseudomonas syringae pathovars using ERIC-, REP-, and IS50-PCR. Journal of Phytopathology, 145, 339–345.
Yamamoto, S., Kasai, H., Arnold, D. L., Jackson, R. W., Vivian, A., & Harayama, S. (2000). Phylogeny of the genus pseudomonas: Intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology, 146, 2385–2394.
Young, J. M. (2010). Taxonomy of Pseudomonas syringae. Journal of plant pathology, 92(1, supplement), S1.5–S1.14.
Zeller, W., Xie, X. L., Bereswill, S., & Geider, K. (1997). Taxonomy and virulence of bacterial blight (pseudomonas syringae pv. Syringae) from pome fruit trees. In K. Rudolph, T. J. Burr, J. W. Mansfield, D. E. Stead, A. Vivian, & J. von Kietzell (Eds.), Pseudomonas syringae Pathovars and related pathogens (pp. 465–469). Dordrecht: Kluwer Academic.
Acknowledgements
This work was supported by the University of Kurdistan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
1. The manuscript has not been published in whole or in part elsewhere;
2. The manuscript is not currently being considered for publication in another journal;
3. The manuscript is not split up into several parts to increase the quantity of submissions;
4. All authors have been personally and actively involved in substantive work leading to the manuscript, and will hold themselves jointly and individually responsible for its content.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Ahmadi, S., Harighi, B. & Abdollahzadeh, J. Phylogenetic relationships of fluorescent Pseudomonad isolates associated with bacterial canker of stone fruit trees in Kurdistan province, Iran. Eur J Plant Pathol 150, 679–689 (2018). https://doi.org/10.1007/s10658-017-1316-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-017-1316-4