
Abstract
Bacterial canker is one of the most important diseases of cherry (Prunus avium). This disease can be caused by two pathovars of Pseudomonas syringae: pv. morsprunorum and pv. syringae. Repetitive DNA polymerase chain reaction-based fingerprinting (rep-PCR) was investigated as a method to distinguish pathovars, races and isolates of P. syringae from sweet and wild cherry. After amplification of total genomic DNA from 87 isolates using the REP (repetitive extragenic palindromic), ERIC (enterobacterial repetitive intergenic consensus) and BOX primers, followed by agarose gel electrophoresis, groups of isolates showed specific patterns of PCR products. Pseudomonas syringae pv. syringae isolates were highly variable. The differences amongst the fingerprints of P. syringae pv. morsprunorum race 1 isolates were small. The patterns of P. syringae pv. morsprunorum race 2 isolates were also very uniform, with one exception, and distinct from the race 1 isolates. rep-PCR is a rapid and simple method to identify isolates of the two races of P. syringae pv. morsprunorum; this method can also assist in the identification of P. syringae pv. syringae isolates, although it cannot replace inoculation on susceptible hosts such as cherry and lilac.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bacterial canker caused by Pseudomonas syringae is an important disease of cherry (Prunus avium) and is one of the major diseases of sweet cherry orchards worldwide. Bacterial canker is also considered to be a major threat for the use of cherry for timber production in farm woodlands in the UK and can cause significant losses in the hardy nursery stock industry (Nicoll, 1993; Vicente, Alves, Russell, & Roberts, 2004). Control of this disease has been limited by a lack of understanding of the taxonomy of the pathogens that cause it, and a lack of reliable and consistent methods of identification and discrimination.
Two pathovars of Pseudomonas syringae can cause bacterial canker in cherry: P. syringae pv. morsprunorum (Psm) and P. syringae pv. syringae (Pss). In the UK, bacterial canker of sweet cherry was considered to be mainly caused by Psm (Burkowicz & Rudolph, 1994; Crosse, 1955; Garrett, Panagopoulos, & Crosse, 1966; Wormald, 1937). A variant of Psm (designated race 2) that showed distinct pathogenicity to some cherry cultivars was identified in East Malling, Kent, UK (Freigoun & Crosse, 1975). More recently, Pss and/or intermediate forms between Psm and Pss were also found in sweet cherry (Garrett & Butler, 1982) and in wild cherry (Garrett & Wood, unpublished; Luz, 1997; Vicente et al., 2004). The disease has been attributed to both pathovars of P. syringae and to intermediate forms in sweet and sour cherry in other European countries (Crosse & Garrett, 1963; Burkowicz & Rudolph, 1994), South Africa (Roos & Hatting, 1986) and USA (Latorre & Jones, 1979).
The different pathovars and races of P. syringae isolates from cherry have been distinguished and characterised by physiological and biochemical tests (Burkowicz & Rudolph, 1994; Garrett et al., 1966; Luz, 1997; Vicente et al., 2004) including the GATTa tests (gelatin liquefaction (G), aesculin hydrolysis (A), tyrosinase activity (T) and tartrate utilisation (Ta)) and the colour of growth in nutrient sucrose broth (Latorre & Jones, 1979; Vicente et al., 2004). Phage typing, the production of syringomycin and serological tests can also be used to increase the speed of detection and discrimination of Psm races and Pss, but these methods do not always distinguish between pathogenic and non-pathogenic Ps isolates isolated from cherry (Garrett et al., 1966; Latorre & Jones, 1979; Vicente et al., 2004). The hypersensitive reaction of tobacco has been used as an indication of pathogenicity (Burkowicz & Rudolph, 1994), but Latorre and Jones (1979) have shown that the results on tobacco do not always correlate well with pathogenicity on cherry. Therefore, inoculation assays in tree branch wounds and leaf scars, fruits, detached twigs and micropropagated plantlets of cherry and lilac have been used to confirm the pathogenicity of isolates (Freigoun & Crosse, 1975; Latorre & Jones, 1979; Vicente & Roberts, 2003; Vicente et al., 2004).
Repetitive sequences present in the genomes of diverse bacterial species have been used to design PCR primers that generate reproducible fingerprints that are useful to assess bacterial diversity at the strain and pathovar level (Louws, Rademaker, & de Bruijn, 1999). The results of Louws, Fulbright, Stephens, and de Bruijn, (1994) indicated that repetitive DNA PCR-based genomic fingerprinting (rep-PCR) with REP (targeting the repetitive extragenic palindromic sequence), ERIC (targeting the enterobacterial repetitive intergenic consensus) and BOX (targeting the DNA sequences of the BOXA subunit of the BOX element of Streptococcus pneumoniae) primers can distinguish isolates of Psm from isolates of Pss. The results of Weingart and Völksch (1997) also showed that REP and ERIC-PCR could differentiate between a Psm isolate from plum and some Pss isolates and that Pss isolates could be highly diverse. Little, Bostock, and Krikpatrick (1998) observed that Pss isolates from stone fruits in California could be separated from Pss isolates from other hosts by ERIC-PCR fingerprinting, and Ménard et al. (2003) differentiated a group of wild cherry isolates from France from Psm races 1 and 2 and other P. syringae isolates using several methods including rep-PCR. After comparing different methods to determine the diversity of fluorescent pseudomonads, Dawson, Fry, and Dancer (2002) concluded that rep-PCR is suitable for the analysis of highly clonal isolates because it is more discriminatory than other DNA fingerprinting techniques and metabolic profiling. The objectives of this work were to assess the usefulness of rep-PCR for discrimination of Psm and Pss isolates from sweet and wild cherry and to characterise a collection of isolates mainly from England, in order to improve understanding of the taxonomy and variation of isolates that cause bacterial canker.
Materials and methods
Bacterial isolates
The bacterial isolates used in this study are presented in Table 1. Most of these isolates originated from sweet and wild cherry trees in England from 1957 to 2000. Five isolates originated from other countries including France (HRI 5355, 5402), Italy (HRI 798), Switzerland (HRI 2928) and New Zealand (HRI 2942). A detailed list of isolates including the source and the year of isolation has been previously published and all isolates have been characterised in biochemical, serological and pathogenicy tests on micropropagated plantlets of wild cherry cv. Charger and accession 1912 and micropropagated plantlets and rooted plants of lilac cv. Sensation (Vicente et al., 2004). The results of some of these tests are summarised in Table 1. The isolates were stored in the WHRI Collection at −76°C (Feltham, Power, Pell, & Sneath, 1978) in liquid medium containing 8 g l−1 of nutrient broth (Difco) and 150 ml l−1 of glycerol.
Isolation of bacterial DNA, PCR conditions and data analysis
Isolates were grown on King’s medium B (KB) (King, Ward, & Raney, 1954) for 24 h at 25°C. Bacterial growth was scraped from the surface of KB plates and suspended in 3 ml of sterile water to produce turbid suspensions that corresponded visually to the McFarland turbidity standards of 3–4 (which correspond to a concentration of 108–109 cells ml−1) (Smibert & Krieg, 1994). Cells from 1.5 ml of these suspensions were harvested by centrifugation for 10 min at 13,000 rpm in a micro-centrifuge. DNA was extracted using the DNeasy tissue extraction kit (Qiagen Ltd, West Sussex, UK) according to the manufacturer’s protocol for extraction of DNA from bacteria and animal tissues. The DNA was extracted from two groups of strains on separate occasions. Three strains (HRI 5260, HRI 5270 and HRI 5275) were included in both groups for comparison. DNA concentration was estimated in a gel by comparison with samples of known concentrations of Lambda DNA.
Primers sequences corresponding to REP1R (5′-IIIICGICGICATCIGGC-3′), REP2I (5′-ICGICTTATCIGGCCTAC-3′), ERIC1R (5′-ATGTAAGCTCCTGGGGATTCAC-3′), ERIC2 (5′-AAGTAAGTGACTGGGGTGAGCG-3′) and BOXA1R (5′-CTACGGCAAGGCGACGCTGACG-3′) described previously (Versalovic, Koeuth, & Lupski, 1991; Versalovic, Schneider, de Bruijn, & Lupski, 1994) were synthesised by Operon Technologies, Inc. (Alameda, California, USA). rep-PCR was carried out as described in Rademaker, Louws, and de Bruijn (1998) with minor modifications including a smaller reaction volume and the addition of MgCl2 to the reaction: PCR reactions were carried out in a total volume of 20 μl containing 1× Gitschier buffer (Kogan, Doherty, & Gitschier, 1987), 3.5 mM MgCl2 for REP and 1.75 mM for ERIC and BOX, 2 μl of dimethylsulfoxide (DMSO) (Fluka, Dorset, UK), 3.2 μg bovine serum albumin (BSA) (Roche, Mannheim, Germany), 40 pmol of each primer (two primers for REP and ERIC and one primer for BOX), 1.25 mM of each of four deoxynucleoside triphosphates, 1.6 U of Taq DNA polymerase (Invitrogen) and approximately 40 ng of template DNA. PCR amplification was performed in a Genius (Techne (Cambridge) Limited, Duxford, Cambridge, UK) thermocycler under the following conditions: one initial cycle at 95°C for 2 min; 30 cycles of denaturation at 94°C for 3 s and 92°C for 30 s, annealing at 40, 52 or 53°C for 1 min with REP, ERIC and BOX primers respectively extension at 65°C for 8 min; single final extension at 65°C for 8 min and then held at 4°C.
Amplified PCR products (6 μl) of each isolate and of controls (with no DNA template included) were separated by gel electrophoresis on 1.2% agarose gels with ethidium bromide at 0.5 μg ml−1 in 0.5× TBE buffer for 8 h at 60 V (1.8 V cm−1) and photographed under UV light. For BOX-PCR amplified products, additional gel electrophoresis was performed with samples diluted 10 and 20 times using the same conditions. A total of six 22-lane gels were run for each set of primers (three gels for each group of DNA extractions). A molecular mass marker (1-kilobase plus DNA ladder, Invitrogen) was loaded on both sides of each gel and the products of three isolates (HRI 5260, HRI 5270 and HRI 5275) corresponding to two different DNA extractions were included in four different gels for comparison.
The rep-PCR fingerprint profiles were used to measure the genetic similarity between isolates. A digital image of each gel was subjected to analysis using the Fingerprinting IITM software (Bio-Rad Laboratories, Hercules, California, USA). Gels were normalised using the standards. In the case of ERIC-PCR, bands of less than 100-bp corresponding to primer-dimer PCR products (also present in the control profiles) were eliminated before analysis. The results of REP-, ERIC- and BOX-PCR genomic fingerprinting were combined using the Fingerprinting IITM software. Cluster analysis of the pairwise similarity values was performed using the Dice similarity coefficient and unweighted pair-group method with arithmetic means (UPGMA) clustering technique.
Results
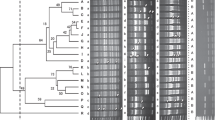
The patterns of the fingerprints of 87 Pseudomonas spp. isolates generated by REP, ERIC and BOX-PCR were complex, with a large number of polymorphic bands. In total, 44 bands were identified in REP (ranging from approximately 150 to 4500 bp), 43 bands in ERIC (ranging from 200 to 5000 bp) and 48 bands in BOX-PCR (ranging from 400 to 3500 bp). The dendrogram obtained from the combined data for all three primer sets is shown in Fig. 1. The combined dendrogram had a cophenetic correlation coefficient of .88. The profiles of the isolates HRI 5260, 5270 and 5275 that were repeated in four different gels had similarities ranging from 89% to 97%.

Dendrogram of genetic similarity of 52 Pseudomonas syringae isolates from wild cherry, 22 from sweet cherry, 13 isolates from other hosts. The similarity is the result of the combined data set of REP, ERIC and BOX primer sets using UPGMA analysis and Dice’s coefficient. The isolates that were repeated in different gels are underlined. The codes to the right of the fingerprints represent the identification according to Vicente et al. (2004), the host of origin (w, wild cherry; s, sweet cherry; p, plum; cl, cherry laurel; l, lilac; pr, pear; my, myrobalan; ph, peach) and the isolate number (Table 1)
At 50% similarity, the Psm isolates of race 1 were separated from all other isolates. At 56% similarity, the other isolates were split into two groups: one group included all the Psm race 2 isolates, two Pseudomonas isolates (HRI 5400 and 5402) from myrobalan and peach and a P. syringae isolate (HRI 7920B) that was not pathogenic on lilac; the second group included all the Pss and other P. syringae isolates. At 65% similarity, 15 Psm race 2 isolates separated from the Psm race 2 isolate HRI 5271, the isolates from myrobablan and peach and the P. syringae isolate HRI 7920B. At this level, three groups of Pss and P. syringae isolates were differentiated: the first group included two Pss isolates (HRI 801 and 2070) from lilac; the second group contained 23 isolates from different hosts, including 22 Pss isolates that were strongly pathogenic on lilac and one P. syringae isolate (HRI 2942) that was not pathogenic on lilac; the third group contained 27 isolates, mainly from wild cherry, including three Pss isolates that were strongly pathogenic (HRI 5841, 7973A and 7928A), 12 Pss isolates that were weakly pathogenic, and 12 P. syringae isolates that were not pathogenic on lilac.
The separate dendrograms (not shown) obtained for each primer set had cophenetic correlation coefficients of .81, .76 and .75 for REP, ERIC and BOX dendrograms respectively. The dendrograms constructed with the independent results of REP and ERIC-PCR clearly separated (at 55 and 53% similarity respectively) the Psm race 1 group and the Psm race 2 group together with the two isolates from myrobalan and peach (HRI 5400, 5402) and a non-pathogenic P. syringae isolate (HRI 7920B) from all other P. syringae isolates. The dendrogram constructed with BOX-PCR results also grouped and separated (at 66% similarity) the Psm race 1 isolates and the Psm race 2 isolates from other P. syringae isolates, but the Psm race 2 isolate HRI 5271 and the two isolates from myrobalan and peach were grouped closer to Psm race 1 than Psm race 2. The groups formed with the Pss and P. syringae isolates in the separate dendrograms were less clear than in the combined dendrogram.
Discussion
All isolates included in this study had been previously characterised by physiological and biochemical tests, serological tests with polyclonal antibodies raised against P. syringae isolates, pathogenicity tests on micropropagated plantlets of lilac cv. Sensation and two wild cherry clones (according to the method developed by Vicente and Roberts (2003)), and pathogenicity tests on rooted plants of lilac (Vicente et al., 2004). Based on these results, the isolates had been identified and grouped (Table 1). The results presented here show that rep-PCR can also differentiate between Psm and Pss and between the two races of Psm and therefore can be an alternative, quicker method to identify and group isolates from cherry and closely related species. Previous studies have also indicated that rep-PCR could be used to differentiate pathovars of P. syringae isolates (Louws et al., 1994; Luz, 1997), but the number of cherry isolates used in these studies was more limited.
The combined dendrogram of REP, ERIC and BOX-PCR fingerprints had a higher cophenetic correlation coefficient than the independent dendrograms, showing that clustering is more accurate and consistent after combining the results. Nevertheless the differentiation of isolates that appear to be more than 90% similar in the combined dendrogram are unreliable because the profiles of the three isolates that originated from different extractions and were loaded in different gels had similarities of 89–97%.
The 16 isolates of Psm race 1 included in this study were more than 92% similar and most of them may be identical despite originating from different Prunus hosts (sweet and wild cherry and plum) and being obtained in years ranging from 1957 to 2001 mainly from southern England, but also including isolates from Switzerland and Italy. Results of the earlier characterisation (Vicente et al., 2004) also indicated that these isolates were very uniform, although the serological results separated two isolates from plum (HRI 2928 and 5300), that were not separated by rep-PCR.
Fifteen out of 16 isolates of Psm race 2 included in this study were more than 88% similar (and many of them may also be identical). These isolates originated from sweet and wild cherry and were obtained from different counties of southern England between 1971 and 2000. The only exception was isolate HRI 5271. This isolate seems to be more similar to the Pseudomonas isolates from myrobalan and peach. Previous results of serological tests also differentiated this isolate from all other Psm race 2 (Vicente et al., 2004). The Psm race 2 isolates and the isolates previously classified as ‘intermediate’ on the basis of biochemical and physiological tests were very uniform in serological and pathogenicity tests. The rep-PCR confirmed this observation and therefore all of these isolates can be included in Psm race 2 as suggested by Vicente et al. (2004). The GATTa tests cannot be used on their own to identify all Psm race 2 isolates; these tests need to be used in conjunction with the colour of growth in nutrient sucrose broth to differentiate the intermediate Psm race 2 isolates from the Pss isolates. On the basis of the biochemical, physiological and serological tests, the Psm race 2 isolates appear to be intermediate forms between Pss and Psm race 1 (Vicente et al., 2004). The results of rep-PCR also support this hypothesis.
In contrast with Psm, the other P. syringae isolates from cherry were highly variable according to the rep-PCR results, but some groups were differentiated. The pathogenicity tests in micropropagated plantlets of cherry and lilac and rooted lilac plants separated these isolates into groups; some of these isolates were not pathogenic in the plants tested and therefore might be non-pathogenic epiphytes although they were isolated from cankers and leaf spots of cherry (Vicente et al., 2004). The serological tests done previously also showed a range of variation in these isolates, but generally these tests did not discriminate between pathogenic, weakly pathogenic and non-pathogenic isolates (Vicente et al., 2004). In contrast, some rep-PCR groups contained mainly strongly pathogenic isolates from different hosts including sweet and wild cherry (that originated mainly from different counties of southern England in years ranging from 1978 to 2000) and also from cherry laurel, plum and pear whilst other rep-PCR groups contained mainly weakly pathogenic or non-pathogenic isolates mainly from wild cherry (that originated from different counties of England between 1995 and 2000). The results of rep-PCR also differentiated the two isolates from lilac from the other isolates from Prunus spp. Nevertheless, there were some exceptions (one non-pathogenic isolate amongst the group of strongly pathogenic isolates and three strongly pathogenic isolates in the group that contained most of the non-pathogenic isolates), so rep-PCR cannot replace pathogenicity tests in susceptible hosts like cherry and lilac for the identification of Pss isolates. The results of rep-PCR support the hypothesis that isolates like HRI 5264 and 7964, that are strongly pathogenic on micropropagated lilac, but less aggressive on micropropagated wild cherry and mature lilac (Vicente & Roberts, 2003; Vicente et al., 2004) might constitute a different variant/race of Pss.
The relative lack of genetic diversity (near identity) of all isolates within each of the two Psm races and diversity of Pss isolates has implications for the epidemiology of the disease and pathogen evolution. The results suggest that Psm is highly adapted and has evolved in a specific association with sweet cherry trees that are generally vegetatively propagated and Pss might be a more recent, opportunistic pathogen that can easily attack different hosts including wild cherry trees that are generally raised from seed and therefore are genetically more variable.
A pathovar is defined as “a strain or set of strains with the same or similar characteristics, differentiated at the infrasubspecific level from other strains of the same species or subspecies on the basis of distinctive pathogenicity to one or more plant hosts” (Dye et al., 1980). The results of Ménard et al. (2003) showed that isolates obtained from wild cherry trees in northern France in 1991 and 1995 were distinct from typical isolates of Psm race 1 and 2. The authors proposed a new pathovar for these isolates (P. syringae pv. avii). The isolates of Ménard et al. (2003) were obtained from wild cherry (which is the same species, P. avium, as sweet cherry, contrary to what is stated by Ménard et al. (2003)) and, like Psm isolates, are not pathogenic on micropropagated lilac, but can cause leaf spots in micropropagated wild cherry plantlets (Vicente et al., unpublished); therefore we consider that the creation of this new pathovar is unnecessary. Although we have identified an isolate (HRI 5271) that seems to be closer to isolates from peach and myrobalan than other Psm, we do not consider the creation of a new pathovar necessary because this isolate was obtained from the same species (P. avium) and can produce leaf spots like other Psm isolates in micropropagated plantlets of cherry.
Cherry trees can have a diverse population of leaf spot and canker-causing bacteria and the isolation and identification of these organisms is not simple. The quickest methods to identify the organisms are possibly serology tests (especially agglutination tests) and/or rep-PCR. The results obtained in REP, ERIC and BOX-PCR can be combined as recommended by Rademaker et al. (2000) to obtain more consistent clustering. In some cases, using PCR with just one or two sets of primers might not be enough to distinguish between isolates of some closely related pathovars: Weingart and Völksch (1997) showed that REP and ERIC fingerprints of a Psm isolate from plum were not easily distinguishable from fingerprints of some isolates of P. savastanoi and Ménard et al. (2003) showed that P. syringae pv. persicae could be distinguished from wild cherry isolates using ERIC and BOX-PCR, but not REP-PCR. Nevertheless, our work shows that the independent results of REP, ERIC and BOX-PCR generally allow the separation of pathovars and races of P. syringae isolates that originated from sweet and wild cherry. Therefore, if time is a limiting factor, PCR with one set of primers should give reliable information; we recommend the use of REP primers because the dendrogram obtained had a higher cophenetic correlation coefficient than the dendrograms obtained with the other two sets of primers. At least one Psm race 1 isolate, two Psm race 2 isolates (one typical and HRI 5271) and four or five Pss isolates representing the main groups identified in this study, should be included in future studies of characterisation and identification of new isolates from cherry. Pathogenicity tests should still be performed especially for the identification of Pss isolates; the inoculation of micropropagated plantlets is a quicker and consistent method that can be used as an alternative to field or twig inoculations (Vicente & Roberts, 2003). Assessing the genetic diversity of the populations that cause bacterial canker should contribute to establish a stable taxonomy and should allow a rational selection of isolates to use in future studies.
References
Burkowicz, A., & Rudolph, K. (1994). Evaluation of pathogenicity and of cultural and biochemical tests for identification of Pseudomonas-syringae pathovars syringae, morsprunorum and persicae from fruit-trees. Journal of Phytopathology, 141, 59–76.
Crosse, J. E. (1955). Bacterial canker of stone-fruits. I. Field observations on the avenues of autumnal infection of cherry. Journal of Horticultural Science, 30, 131–142.
Crosse, J. E., & Garrett, C. M. E. (1963). Studies of the bacteriophagy of Pseudomonas mors-prunorum, Ps. syringae and related organisms. Journal of Applied Bacteriology, 26, 159–177.
Dawson, S. L., Fry, J. C., & Dancer, B. N. (2002). A comparative evaluation of five typing techniques for determining the diversity of fluorescent pseudomonads. Journal of Microbiological Methods, 50, 9–22.
Dye, D. W., Bradbury, J. F., Goto, M., Hayward, A. C., Lelliott, R. A., & Schroth, M. N. (1980). International standards for naming pathovars of phytopathogenic bacteria and a list of pathovar names and pathotype strains. Review of Plant Pathology, 59, 153–168.
Feltham, R. K. A., Power, A. K., Pell, P. A., & Sneath, P. H. A. (1978). A simple method for the storage of bacteria at −76°C. Journal of Applied Bacteriology, 44, 313–316.
Freigoun, S. O., & Crosse, J. E. (1975). Host relations and distribution of a physiological and pathological variant of Pseudomonas morsprunorum. Annals of Applied Biology, 81, 317–330.
Garrett, C. M. E., & Butler, M. (1982). Virulence of isolates from cherry. East Malling Research Station Report for 1981, 74.
Garrett, C. M. E., Panagopoulos, C. G., & Crosse, J. E. (1966). Comparison of plant pathogenic Pseudomonads from fruit trees. Journal of Applied Bacteriology, 29, 342–356.
King, E. O., Ward, M. K., & Raney, D. R. (1954). Two simple media for the demonstration of pyocyanin and fluorescein. Journal of Laboratory and Clinical Medicine, 44, 301–307.
Kogan, S. C., Doherty, M., & Gitschier, J. (1987). An improved method for prenatal-diagnosis of genetic-diseases by analysis of amplified DNA-sequences—application to hemophilia-A. New England Journal of Medicine, 317, 985–990.
Latorre, B. A., & Jones, A. L. (1979). Pseudomonas morsprunorum, the cause of bacterial canker of sour cherry in Michigan, and its epiphytic association with P. syringae. Phytopathology, 69, 335–339.
Little, E. L., Bostock, R. M., & Kirkpatrick, B. C. (1998). Genetic characterization of Pseudomonas syringae pv. syringae strains from stone fruits in California. Applied and Environmental Microbiology, 64, 3818–3823.
Louws, F. J., Fulbright, D. W., Stephens, C. T., & de Bruijn, F. J. (1994). Specific genomic fingerprints of phytopathogenic Xanthomonas and Pseudomonas pathovars and strains generated with repetitive sequences and PCR. Applied and Environmental Microbiology, 60, 2286–2295.
Louws, F. J., Rademaker, J. L. W., & de Bruijn, F. J. (1999). The three Ds of PCR-based genomic analysis of phytobacteria: Diversity, detection, and disease diagnosis. Annual Review of Phytopathology, 37, 81–125.
Luz, J. P. M. (1997). Detection and epidemiology of bacterial canker (Pseudomonas syringae) on wild cherry (Prunus avium), PhD thesis. University of Reading, UK.
Ménard, M., Sutra, L., Luisetti, J., Prunier, J. P., & Gardan, L. (2003). Pseudomonas syringae pv. avii (pv. nov.), the causal agent of bacterial canker of wild cherries (Prunus avium) in France. European Journal of Plant Pathology, 109, 565–576.
Nicoll, F. J. (1993). Genetic improvement of cherry for farm woodlands. Quarterly Journal of Forestry, 87, 187–194.
Rademaker, J. L. W., Hoste, B., Louws, F. J., Kersters, K., Swings, J., Vauterin, L., Vauterin, P., & de Bruijn, F. J. (2000). Comparison of AFLP and rep-PCR genomic fingerprinting with DNA—DNA homology studies: Xanthomonas as a model system. International Journal of Systematic and Evolutionary Microbiology, 50, 665–677.
Rademaker, J. L. W., Louws, F. J., & de Bruijn, F. J. (1998). Characterization of the diversity of ecologically important microbes by rep-PCR genomic fingerprinting. In: de Bruijn F. J. (Ed.) Molecular Microbial Ecology Manual. 3.4.3. (pp. 1–27). Dordrecht, the Netherlands: Kluwer Academic Publishers.
Roos, I. M. M., & Hatting, M. J. (1986). Bacterial canker of sweet cherry in South Africa. Phytophylactica, 18, 1–4.
Smibert, R. M., & Krieg, N. R. (1994). Phenotypic characterization. In: Gerhard, Murray, Wood, & Krieg (Eds.) Methods for general and molecular bacteriology (pp. 607–654). Washington DC: American Society for Microbiology.
Versalovic, J., Koeuth, T., & Lupski, J. R. (1991). Distribution of repetitive DNA-sequences in eubacteria and application to fingerprinting of bacterial genomes. Nucleic Acids Research, 19, 6823–6831.
Versalovic, J., Schneider, M., de Bruijn, F. J., & Lupski, J. R. (1994). Genomic fingerprinting of bacteria using repetitive sequence-based polymerase chain reaction. Methods in Molecular and Cellular Biology, 5, 25–40.
Vicente, J. G., Alves, J. P., Russell, K., & Roberts, S. J. (2004). Identification and discrimination of Pseudomonas syringae isolates from wild cherry in England. European Journal of Plant Pathology, 110, 337–351.
Vicente, J. G., & Roberts, S. J. (2003). Screening wild cherry micropropagated plantlets for resistance to bacterial canker. In: Iacobellis N. S., et al. (Eds.) Pseudomonas syringae and related pathogens: Biology and genetic (pp. 467–474). Dordrecht, the Netherlands: Kluwer Academic Publishers.
Weingart, H., & Völksch, B. (1997). Genetic fingerprinting of Pseudomonas syringae pathovars using ERIC-, REP-, and IS50-PCR. Journal of Phytopathology, 145, 339–345.
Wormald, H. (1937). Bacteriosis of stone fruit trees in Britain. VI. Field observations on bacteriosis of sweet cherry trees. The Journal of Pomology and Horticultural Science, 15, 35–48.
Acknowledgements
This work was carried out under a plant health licence from and funded by the Department for Environment, Food and Rural Affairs (Defra) of the UK. We thank Shazia Akram and João Pedro Alves for laboratory assistance. We also thank Karen Russell (East Malling Research, UK) for assistance in obtaining isolates and Constance M.E. Garrett, Christine Lewis (East Malling Research, UK) and João P. Luz (University of Reading, UK) for supplying some of their isolates. We thank Brian Keenan from Bio-Rad Laboratories Ltd for the use of FingerprintingTM II.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vicente, J.G., Roberts, S.J. Discrimination of Pseudomonas syringae isolates from sweet and wild cherry using rep-PCR. Eur J Plant Pathol 117, 383–392 (2007). https://doi.org/10.1007/s10658-007-9107-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-007-9107-y