
Abstract
Banana bunchy top disease is caused by the Banana bunchy top virus (BBTV), which is a species of the genus Babuvirus in the family Nanoviridae. In this study, we collected 61 isolates of BBTV from Sumatra Island. The sequences of DNA-R and DNA-S of 61 samples and DNA-U3 of 37 samples were determined. Although the Sumatra population has different sequences in the stem-loop region in DNA-U3, it is generally very similar, with little or no variation according to planting area or the varieties and genome types of the host. The comparison of sequences between Sumatra isolates and those from other areas showed that the sequence identity with the Asian group is higher than with the South Pacific group. The genetic analysis showed that the Sumatra population has very low diversity compared with other areas. Recombination analysis revealed that there was no evidence of recombination in the Sumatra population. The selection pressure analysis indicated that all Sumatra populations received negative selection. The selection pressure among the viruses infecting the banana varieties having the genotypes of AAB or ABB received a slightly weaker negative selection than that of the viruses infecting the varieties with AA or AAA genotypes. A neutrality test suggested the sudden expansion of the West Sumatra population. This is the first report to survey the BBTV on Sumatra Island.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Banana is one of the most important crops in many tropical and subtropical areas as both a food and cash crop. Indonesia is the world’s sixth largest producer of bananas (FAOSTAT 2011). However, the banana plantations in Indonesia are currently under serious threat from banana bunchy top disease (BBTD).
BBTD is caused by Banana bunchy top virus (BBTV), which is the type species of the genus Babuvirus in the family Nanoviridae, and is transmitted by aphids (Pentalonia nigronervosa) (Dale 1987). BBTV causes symptoms of plant stunting, leaf chlorosis, and leaf blight on bananas (Fig. S1). The genome of BBTV consists of six circular single-strand DNA components named DNA-R, -U3, -S, -M, -C, and -N, each approximately 1.1 kb, that are individually encapsidated within separate icosahedral virions, each 18–20 nm in diameter. DNA-R encodes replication initiation protein (M-Rep), DNA-S encodes a coat protein (CP), DNA-M encodes a movement protein (MP), DNA-C encodes a cell cycle link protein (Clink), and DNA-N encodes a nuclear shuttle protein (NSP); the function of the DNA-U3 encoded protein, however, is unknown (Vetten et al. 2012). BBTV is divided into two groups, named the Asian group and South Pacific group based on phylogenetic analysis of all components (Karan et al. 1994; Wanitchakorn et al. 2000; Vishnoi et al. 2009).
BBTV has two conserved sequences, named the stem-loop common region (CR-SL) and the major common region (CR-M), in all genome components (Harding et al. 1993; Burns et al. 1995). The CR-SL is important for replication and contains a stem-loop structure (TATTATTAC) and an “iterated sequence” (F1, F2, and R) region (Herrera-Valencia et al. 2006). The CR-M is known to be a GC-rich region, but its function is not clear (Burns et al. 1995).
Until now, in Indonesia, BBTV has only been identified on the Java and Bali Islands, and it was determined that the virus there belonged to the Asian group based on phylogenetic analysis of DNA-R (Furuya et al. 2004; Pinili et al. 2011). Sumatra Island is one of the main banana-producing areas, but it had not been surveyed for BBTV. In this study, we surveyed banana crops on Sumatra to assess whether they were infected with BBTV. Subsequently, we determined the nucleotide sequence of the DNA-R, -U3, and -S components, and analyzed them phylogenetically. In addition, the genetic structure and diversity of the Sumatra population was analyzed.
Materials and methods
Collection of banana samples
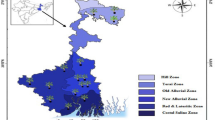
From autumn 2010 to summer 2012, we collected 61 leaf samples of banana plants showing typical symptoms of BBTD from four locations in West Sumatra, three locations each in North Sumatra, Riau and Lampung, and one location in South Sumatra (Fig. 1). The banana leaf samples were dehydrated using silica gel and imported with the permission of the Ministry of Agriculture, Forestry and Fisheries, Japan. The banana varieties and genome type of the collected samples are listed in Table S1.

Banana leaf sampling locations for the detection of BBTV
DNA extraction and detection of BBTV by PCR
Total DNA was extracted from 0.01 g of dehydrated leaf samples using a DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). Primers for the amplification of full length DNA-R, DNA-U3, and DNA-S were used as described in Table S2 (Karan et al. 1994; Furuya et al. 2004; Zhuang and Zhi-Xin 2005). DNA-R and DNA-S were amplified by PCR using Ex Taq polymerase (TaKaRa, Tokyo, Japan), and DNA-U3 was amplified by PCR using Platinum® Pfx DNA Polymerase (Invitrogen, Carlsbad, CA). The PCR conditions consisted of 1 cycle of 94 °C for 2 min, followed by 40 cycles of 94 °C for 1 min, the annealing temperature of each primer pair for 1 min, and 72 °C (Ex Taq) or 68 °C (Platinum® Pfx) for 2 min, and a final extension for 10 min at 72 °C (Ex Taq) or 68 °C (Platinum® Pfx).
Previous studies showed that the phylogenetic trees deduced from the sequences of all six DNA components showed the same hereditary patterns across individual isolates (Vishnoi et al. 2009; Stainton et al. 2012). Moreover, since DNA-R (encoding Rep) and DNA-S (encoding CP) were important for the multiplication of viral genomes and the formation of a virion, respectively and DNA-U3 is known to have high diversity compared with other components, we used DNA-R, -U3, and -S for genetic analysis in this study.
Cloning and sequencing
PCR products were separated by electrophoresis with 1 % agarose gel and purified using a Fast Gene Gel/PCR Extraction Kit (Genetics, Tokyo, Japan). The purified DNA-R and DNA-S fragments were cloned into pMD20 vector (TaKaRa), and the DNA-U3 fragments were cloned into pBluescript SK(+) (Agilent Technologies, Palo Alto, CA) using T4DNA Ligase (TaKaRa) after digestion with the restriction enzyme Sma I. These vectors were then transformed into Escherichia coli JM109. Positive clones were selected by blue/white screening. Subsequently, plasmid DNA was extracted using an Illustra plasmid Prep Mini Spin kit (GE Healthcare Life Sciences, Buckinghamshire, UK).
The nucleotide sequence was determined using an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA). At least three clones of each PCR fragment were sequenced. The secondary structure of the stem-loop region in DNA-U3 was estimated by the mfold Web Server (Zugker 2003).
Phylogenetic analysis of DNA-R, DNA-U3 and DNA-S
Sequences of each component of BBTV Sumatra isolates, the isolates of other areas, and Abaca bunchy top virus, used as an outgroup, were aligned by ClustalW in MEGA5 (Tamura et al. 2011). Each component of the phylogenetic trees was constructed with MEGA5 using the Neighbor-joining (NJ) method with 1000 bootstrap replications (Saitou and Nei 1987). Kimura’s two parameter method was applied for the NJ method (Kimura 1980). The generated trees were displayed by TreeExplorer in MEGA5.
Recombination analysis
Putative recombination breakpoints in all sequences were identified using RDP (Martin and Rybicki 2000), GENECONV (Padidam et al. 1999), BOOTSCAN (Salminen et al. 1995), MAXCHI (Smith 1992), CHIMAERA (Posada and Crandall 2001), SISCAN (Gibbs et al. 2000) and 3SEQ (Boni et al. 2007) methods implemented in the program RDP4 (Martin et al. 2010).
Estimation of genetic diversity and selection pressure
Sequence identity analysis within and between Sumatra isolates and the isolates of other areas was performed using a Fasta Homology Search in GENETYX ver. 11 (Genetyx Corp., Tokyo, Japan). To assay the genetic diversity of the Sumatra population and other populations, we calculated the average of genetic distance. The average genetic distance of each component was calculated by MEGA5 using the maximum composite likelihood (MCL) method and expressed as the average number of nt substitutions per site in each pair of sequences (Tamura et al. 2004).
The selection pressure was estimated by the dN/dS ratio. The value of dN represents the average number of non-synonymous substitutions per non-synonymous site, and dS is the average number of synonymous substitutions per synonymous site. The dN and dS values were estimated separately based on the Pamilo-Bianchi-Li method (Li 1993; Pamilo and Bianchi 1993).
Population demography
The software package DNASP v5 (Librado and Rozas 2009) was used to perform the Tajima’s D (Tajima 1989) and Fu and Li’s D and F (Fu and Li 1993) statistical tests and to estimate the haplotype diversity. Tajima’s D is based on the difference between the number of segregating sites and the average number of nt differences. Fu and Li’s D test is based on the difference between the numbers of singletons and the total numbers of mutations. Fu and Li’s F test is based on the difference between the numbers of singletons and the average number of nt differences between each pair of sequences. Haplotype diversity was calculated based on the frequency and number of haplotypes in the population.
Results and discussion
Sequences of the individual components
DNA-R and DNA-S were detected from all samples. However DNA-U3 was detected from only 37 samples. From the remaining 24 samples, we could not obtain sufficient DNA-U3 despite several attempts. The inability to obtain sufficient template may be because DNA extraction was performed from dried, rather than fresh leaves, but the detailed reason is unclear. So, in this study, we analyzed DNA-U3 of 37 isolates.
All DNA-R components of BBTV Sumatra isolates contained 1104 nt and an ORF coding M-Rep was observed. All DNA-S components contained 1058 nt and an ORF coding CP was observed. Most of the DNA-U3 components contained 1036 nt. However, some contained 1037 nt. Previous studies showed that the position of the start codon of ORF encoded by DNA-U3 was UUG instead of AUG as observed in some Chinese isolates, and that all isolates have a smaller putative ORF with a GUG start codon located downstream (Yu et al. 2012). Since all Sumatra isolates had a UUG codon in position 143–145 instead of AUG as in an Australian and some Chinese isolates, GUG could be considered as the start codon. However, the length of the ORF starting from GUG was much longer than that of some of the Chinese isolates, and the Poly A signal (Beetham et al. 1999) was included in this region. Therefore, although it is a rare phenomenon in the genome of a virus, we estimated that the UUG is a start codon in the DNA-U3 of Sumatra isolates. Thus, an unknown ORF-encoded protein was estimated at 143–280 nt.
BBTV has two conserved sequences, named the stem-loop common region (CR-SL) and the major common region (CR-M), in all components (Harding et al. 1993; Burns et al. 1995). In the CR-SL, the stem-loop structure was observed with the nonanucleotide sequence “TATTATTAC”. Interestingly, there were two types of sequences in the stem-loop region of CR-SL in the Sumatra isolates, named Type A and Type B (Fig. 2). However, the secondary structure of the stem-loop was maintained in both types, and the nonanucleotide sequence was not changed. Thus, it is believed that although a sequence difference was observed, there is no difference in the function of the stem-loop. Most importantly, only the South areal isolates had the Type B sequence in the CR-SL. Thus the South areal isolates might have evolved independently and been adapted by geographical isolation.

The secondary structure of the stem-loop region of DNA-U3 in BBTV Sumatra isolates. Sequence differences are indicated with boxes
Sequence identity and phylogenetic analysis
The sequence identity among and within each location was very high regardless of the banana variety and location. Sumatra isolates have a high identity to isolates of the Asian group (Table S3). Previous studies have shown that the nucleotide and amino acid sequences of DNA-R and DNA-S are generally conserved at a high level (Stainton et al. 2012). Similarly, the sequence identity of DNA-R and DNA-S on Sumatra Island were more than 98 and 99 %, respectively. On the other hand, DNA-U3 was notable for having the highest diversity of all BBTV components (Hyder 2009), but the DNA-U3 of Sumatra isolates had high identity (Table S3). This is similar to the results for the Pakistan and Tonga populations (Hyder et al. 2011; Stainton et al. 2012). Since the nucleotide sequence of DNA-U3 had not been determined in Indonesia isolates until now, our data will be especially useful for the investigation of BBTV in Indonesia. To assay the genetic diversity of the BBTV population, we estimated the average nt distance for each component. The results of the genetic diversity analysis indicated that the BBTV population on Sumatra Island has very low diversity, even when each component is used, compared to the populations in other countries (Table 1).
The results of the phylogenetic analysis based on the nucleotide sequences of DNA-R, -S and -U3 are shown in Fig. 3 and Fig S2. These results showed that the Sumatra isolates belonged to the Asian group. Furthermore, in the Asian group, isolates from Indonesia, Japan, the Philippines and Taiwan were closer to the Sumatra isolates than were those from China and Vietnam.

Phylogenetic analysis of BBTV Sumatra isolates and the isolates of other regions based on the full length of nucleotide sequences of DNA-R. The nucleotide sequences of ABTV are used as an outgroup. The number of each node indicates a bootstrap percentage based on 1000 replications (only values >75 % are shown) in NJ
Recombination analysis
In the recombination analysis, although some of programs identified a recombination breakpoint, the support for this result was insufficient, and we judged that recombination had not occurred in the Sumatra BBTV population. Thus, although there were two types of sequences in the stem-loop region of DNA-U3 in the Sumatra population, it was suggested that these mutations were not the result of recombination, but rather were natural mutations. Therefore, it was suggested that there is only one population of BBTV existing on Sumatra Island. Thus, on Sumatra Island, it would be possible to purposefully advance the selection of a banana variety resistant to this one BBTV genotype. That such a promising resistant variety has not yet been found is probably the reason why the gene of the virus is even now stable without tending toward diversity.
Genetic diversity and selection pressure in individual areas of Sumatra Island
To assay the genetic diversity of the BBTV population, we estimated the average nt distance for each component in every area. The results of the genetic diversity analysis indicated that the BBTV population of the South area has higher diversity than the other areas (Table 2). We also estimated the pattern of selective constraint on the ORF of DNA-R and DNA-S by analyzing the dN/dS ratio. The dN values for all the encoding regions were smaller than the dS values, and the dN/dS ratios of both components were <1 (Table 2). However, the dN/dS ratio was different between the two components. In DNA-R, the Riau and South areal populations received strongly negative selection compared with the other areas. However, in the case of DNA-S, the West Sumatra, Riau and North Sumatra populations received strongly negative selection compared with the South areal population. This suggests that the degree of selection pressure differs depending on the area and component. Especially in DNA-S, most of the substitutions were synonymous substitutions. We consider that, since the role of CP is critical for the survival of the virus in the host plant, the sequence of DNA-S is conserved among all areas.
We also investigated whether the genetic diversity and selective pressure differed among the different banana genome types. These results showed that viruses infecting the banana varieties having the AAB or ABB genotypes had slightly higher diversity than those infecting the varieties with AA or AAA genotypes (Table 3). The selection pressure of DNA-R was almost the same. However, the dN/dS value of the DNA-S of virus infecting the banana varieties having the AAB or ABB genotype was higher than those of the virus infecting the AA or AAA variety. As for the reason underlying this phenomenon, BBTV may adapt banana varieties that include a B genotype. Previous studies have shown that some banana varieties including the B genotype have resistance for BBTV (Hooks et al. 2009; Furuya et al. 2012). Perhaps, since there may have been some resistant bananas in our sample, the viruses infecting banana varieties including the B type genotype may have greater diversity than those infecting banana varieties including only the A genotype.
Neutrality test
We estimated the nt polymorphism of each population using their DNA-R and DNA-S sequences by the statistical tests Tajima’s D and Fu and Li’s D and F in order to discriminate between demographic expansion and contraction (Table 4). A negative value from these statistical tests indicates that the population is in a state of increase, while a positive value indicates a decrease in the population size or that there is a balancing selection. The Tajima’s D, and Fu and Li’s D and F values for all populations were negative, but the P-values for the populations of North Sumatra and South Sumatra were both >0.10, and the results were not conclusive in these cases. In DNA-R of the Riau population, the P-value was 0.1 > P > 0.05, however in DNA-S, the P-value was P > 0.10. Therefore, although we can not judge the state of the Riau population precisely, the Riau population may be increasing in size. On the other hand, the P-values for the West Sumatra population were P < 0.01 or P < 0.02, suggesting that this group is increasing in size. Since West Sumatra was the most recent location to adopt the commercial cultivation of the banana, BBTV may have adapted to banana in West Sumatra and may be growing at the present time. Since many varieties of banana which were cultivated in West Sumatra were cultivated in other region as well, this phenomenon must be caused by the effects of some unknown environmental reasons rather than difference of varieties of banana.
In this study, we detected BBTV on Sumatra Island for the first time. The sequences data of BBTV obtained in our investigation will be useful as fundamental knowledge for the future control of BBTD in Indonesia.
References
Beetham, P. R., Harding, R. M., & Dale, J. L. (1999). Banana bunchy top virus DNA-2 to 6 are monocistronic. Archives of Virology, 144, 89–105.
Boni, M. F., Posada, D., & Feldman, M. W. (2007). An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics, 176, 1035–1047.
Burns, T. M., Harding, R. M., & Burns, J. L. (1995). The genome organization of banana bunchy top virus: analysis of six ssDNA components. Journal of General Virology, 76, 1471–1482.
Dale, J. L. (1987). Banana bunchy top: an economically important tropical plant virus disease. Advances in Virus Research, 33, 301–325.
F.A.O.S.T.A.T. Anonymous. (2011). ProdSTAT: Crop. UN Food and Agriculture Organization, p.567 http://faostat.fao.org/site/567/DesktopDefault.aspx?PageID=567. Accessed date 27 Aug 2013.
Fu, Y. X., & Li, W. H. (1993). Statistical tests of neutrality of mutations. Genetics, 133, 693–709.
Furuya, N., Somowiyarjo, S., & Natsuaki, K. T. (2004). Virus detection from local banana cultivars and the first molecular characterization of Banana bunchy top virus in Indonesia. Journal of Agricultural Science, Tokyo University of Agriculture, 49, 75–81.
Furuya, N., Kawano, S., & Natsuaki, K. T. (2012). Evaluation of resistance of Japanese fibre banana cv. Itobasho (Musa balbisiana var. liukiuensis) to Banana bunchy top virus. Japanese Journal of Phytopathology, 78, 108–110.
Gibbs, M. J., Armstrong, J. S., & Gibbs, A. J. (2000). Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics, 16, 573–582.
Harding, M. R., Burns, T. M., Hafner, G. J., Dietzgen, R. G., & Dale, J. L. (1993). Nucleotide sequence of one component of the banana bunchy top virus genome contains a putative replicase gene. Journal of General Virology, 74, 323–328.
Herrera-Valencia, V. A., Dugdale, B., Harding, R. M., & Dale, J. L. (2006). An iterated sequence in the genome of Banana bunchy top virus is essential for efficient replication. Journal of General Virology, 87, 3409–3412.
Hooks, C. R. R., Manandhar, R., Perez, E. P., Wang, K. H., & Almeida, R. P. P. (2009). Comparative susceptibility of two banana cultivars to Banana Bunchy Top Virus under the laboratory and field environments. Journal of Economic Entomology, 102, 897–904.
Hyder, M. Z. (2009). Sequencing and Genetic Characterization of Major DNA Components of Banana Bunchy Top Virus. Pir Mehr Ali Shah, Arid Agriculture University Rwalpindi, Pakistan, Ph.D. Thesis.
Hyder, M. Z., Shah, S. H., Hameed, S., & Naqvi, S. M. S. (2011). Evidence of recombination in the Banana bunchy top virus genome. Infection, Genetics and Evolution, 11, 1293–1300.
Karan, M., Harding, R. M., & Dale, J. L. (1994). Evidence for two groups of banana bunchy top virus isolates. Journal of General Virology, 75, 3541–3546.
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution, 16, 111–120.
Li, W. H. (1993). Unbiased estimation of the rates of synonymous and nonsynonymous substitution. Journal of Molecular Evolution, 36, 96–99.
Librado, P., & Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451–1452.
Martin, D., & Rybicki, E. (2000). RDP: detection of recombination amongst aligned sequences. Bioinformatics, 16, 562–563.
Martin, D. P., Lemey, P., Lott, M., Moulton, V., & Posada, D. (2010). RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics, 26, 2462–2463.
Padidam, M., Sawyer, S., & Fauquet, C. M. (1999). Possible emergence of new geminiviruses by frequent recombination. Virology, 265, 218–225.
Pamilo, P., & Bianchi, N. O. (1993). Evolution of the Zfx and Zfy genes: rates and interdependence between the genes. Molecular Biology and Evolution, 10, 271–281.
Pinili, M. S., Nyana, D. N., Suastika, G., & Natsuaki, T. K. (2011). Molecular analysis of Banana bunchy top virus first isolated in Bali, Indonesia. Journal of Agricultural Science, Tokyo University of Agriculture, 56, 125–134.
Posada, D., & Crandall, K. A. (2001). Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proceedings of the National Academy of Sciences of the United States of America, 98, 13757–13762.
Saitou, N., & Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4, 406–425.
Salminen, M. O., Carr, J. K., Burke, D. S., & McCutchan, F. E. (1995). Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Research and Human Retroviruses, 11, 1423–1425.
Smith, J. M. (1992). Analyzing the mosaic structure of genes. Journal of Molecular Evolution, 34, 126–129.
Stainton, D., Kraberger, S., Walters, M., Witshire, E. J., Rosario, K., Halafihi, M., Lolohea, S., Katoa, I., Faitua, T. H., Aholelei, W., Taufa, L., Thomas, J. E., Collongs, D. A., Martin, D. P., & Varsani, A. (2012). Evidence of inter-component recombination, intra-component recombination and reassortment in banana bunchy top virus. Journal of General Virology, 93, 1103–1119.
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123, 585–595.
Tamura, K., Nei, M., & Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proceedings of the National Academy of Sciences of the United States of America, 101, 11030–11035.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., & Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739.
Vetten, H. J., Grigoras, I., Harding, R., Randles, J. W., Sano, Y., Thomas, J. E., Timchenko, T., & Yeh, H. H. (2012). In virus taxonomy—Ninth report of the international committee on taxonomy of viruses. In A. M. Q. King, M. J. Adams, E. B. Carstens, & E. J. Lefkowitz (Eds.), Babuvirus (pp. 400–404). San Diego: Associated Press, Elsevier Inc.
Vishnoi, R., Raj, S. K., & Prasad, V. (2009). Molecular characterization of an Indian isolate of Banana bunchy top virus based on six genomic DNA components. Virus Genes, 38, 334–344.
Wanitchakorn, R., Harding, R. M., & Dale, J. L. (2000). Sequence variability in the coat protein gene of two groups of banana bunchy top isolates. Archives of Virology, 145, 593–602.
Yu, N. T., Zhang, Y. L., Feng, T. C., Wang, J. H., Kulye, M., Yang, W. J., Lin, Z. S., Xiong, Z., & Liu, Z. X. (2012). Cloning and sequence analysis of two banana bunchy top virus genomes in Hainan. Virus Genes, 44, 488–494.
Zhuang, T. E., & Zhi-Xin, L. (2005). Cloning and sequencing of DNA components of Banana bunchy top virus Hainan isolate. Chinese Journal of Agricultural Biotechnology, 2, 91–97.
Zugker, M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research, 31, 3406–3415.
Acknowledgments
We thank Professor Kazusato Ohshima (Faculty of Agriculture, Saga University) who provided constructive suggestions to improve the manuscript.
Conflict of interest
The authors have no conflict of interest directly relevant to the content of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 499 kb)
Rights and permissions
About this article

Cite this article
Chiaki, Y., Nasir, N., Herwina, H. et al. Genetic structure and diversity of the Banana bunchy top virus population on Sumatra Island, Indonesia. Eur J Plant Pathol 143, 113–122 (2015). https://doi.org/10.1007/s10658-015-0669-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-015-0669-9