
Abstract
Identification of the spatial extent of genetic structuring that may be influenced by evolutionary, ecological and historical factors is critical for effective conservation or management strategies. Masu salmon Oncorhynchus masou is commonly distributed in Far East, however, many local populations have been under threats of decline due to habitat destruction, overexploitation, and genetic introgression. To reveal the spatial genetic structure of native masu salmon populations in Hokkaido, masu salmon samples were collected from 16 rivers in which there was no official record of artificial releases of any masu salmon stock and were analyzed using 15 microsatellite loci. A Bayesian assignment test revealed that masu salmon populations were divided into two genetically distinct groups: the northeastern and southwestern groups. For within-group genetic structure, all populations, except for geographically proximate populations, were significantly different from each other. AMOVA revealed that genetic variation at among-group level based on groups identified assignment test was greater than that of groups based on geographic locations. There was no significant IBD for the 16 populations. However, the Mantel test revealed significant IBD for the northeastern group, but did not for the southwestern group. This study suggested that native masu salmon populations in Hokkaido exhibit a hierarchical genetic structure that is largely a result of their precise homing behavior. The results of this study also highlight the importance of defining populations by using genetic data rather than by using predefined populations based on geographic locations for the correct determination of genetic structure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Patterns of genetic differentiation are influenced by evolutionary, ecological, and historical factors experienced by organisms, and thus identification of the spatial extent of population genetic structuring and the number of such groups or units is critical for effective conservation or management strategies (Moritz 1994; Frankham et al. 2004; Waples and Gaggiotti 2006). However, genetic structures of many aquatic organisms have also been affected by environmental changes including natural and anthropogenic alterations (Caputo et al. 2004; Yamamoto et al. 2004; Kitanishi et al. 2017a). For instance, loss of genetic diversity and homogenization of genetic features caused by artificial releases of non-natives or domesticated fish have been reported worldwide (e.g., Eldridge and Naish 2007; Glover et al. 2012), suggesting that conservation of indigenous genetic features within local populations is urgent issue for maintain genetic diversity. A critical first step is to identify the extent of genetic structure of native populations as baseline information for the effective conservation and for estimation of the artificial effects on genetic structuring (Allendorf et al. 2001; Palsbøll et al. 2007).
Masu salmon Oncorhynchus masou is distributed in Russia, Korea, Taiwan, and Japan surrounding the Sea of Japan and the Sea of Okhotsk. Although masu salmon are common in this area, many local populations, including its subspecies such as Biwa salmon (O. masou subsp.) and red-spotted masu salmon (O. m. ishikawae), have been under threats of decline due to habitat destruction, overexploitation, and genetic introgression (Fukushima et al. 2007; Kawamura et al. 2007; Tamate and Hayajiri 2008). Like other salmonids, masu salmon exhibits two life histories: anadromous and resident (Kato 1991). In Hokkaido, all females are anadromous, whereas males can be either anadromous or resident and the ratio of resident to anadromous males decreases with increasing latitude (Kato 1991). After downstream migration, anadromous fish move into the Sea of Okhotsk along the Tsushima Current or Pacific coast of southern Hokkaido to feed. After one year of sea life, they return to their natal river to spawn (i.e., natal homing, Kato 1991), leading to the development of distinct populations within local spawning areas (Kitanishi et al. 2009). Because of the importance of masu salmon as a fisheries resource and the reduction of commercial catches over the past few decades (Miyakoshi et al. 2004), hatchery-reared fish have been released over a wide area in Japan for resource enhancement (Morita et al. 2006). Although loss of genetic diversity (Edpalina et al. 2004) and abnormal behavior (Yamamoto and Reinhardt 2003) of domesticated masu salmon have been reported, insufficient attention has been directed toward artificial releases of the stock populations and/or recipient populations. Consequently, the losses of genetic diversity and genetic introgressions by the non-native stocks have occurred throughout Japan, especially in Honshu Islands, main island of Japan, and indigenous genetic features and genetic structuring of many local populations have been lost due to the genetic introgressions (Kawamura et al. 2007; Kitanishi et al. 2017b).
Previous studies focused on the genetic structure of masu salmon showed relatively high levels of genetic differentiation among populations and reported the existence of regional genetic groups in Hokkaido (Okazaki 1986; Suzuki et al. 2000; Kitanishi et al. 2007; Yu et al. 2010, 2012). However, the number of groups detected and determination of groups differed among the studies. One possible reason for the discordance among studies is that most studies analyzed populations in which a vast number of hatchery-reared stocks were released annually or wild populations in which hatchery stock had been released in the past, suggesting that the studies included the influence of hatchery-reared fish rather than analyzing purely native populations, leading to the ambiguity or homogenization in the genetic structuring (Eldridge and Naish 2007). In addition, given the precise homing of masu salmon (Miyakoshi et al. 2012; Kitanishi et al. 2017c), it is likely that each river or regional population potentially has indigenous genetic characteristics, and that additional genetic structuring exists in masu salmon populations.
The aim of this study is to provide baseline information on genetic structuring for planning management and conservation strategies of masu salmon by investigating the genetic structure of masu salmon populations in Hokkaido. This study assessed spatial genetic structuring of 16 native populations in which there was no official record of artificial releases by using 15 microsatellite loci. In addition, a prior definition of regional populations based on geographic locations (e.g., Pacific, Sea of Okhotsk, and Sea of Japan in the study area) has been typically used in empirical studies. However, several model-based approaches that could infer the number of genetic groups have been introduced (e.g., Pritchard et al. 2000; Corander et al. 2004), and a discrepancy between predefined populations and spatial genetic structure using solely genetic data has been reported for other salmonid species (e.g., Vähä et al. 2007). Therefore, to evaluate the usefulness of these two methods (i.e. genetic data vs. geographic location), a Bayesian assignment test was used to infer population genetic structure in Hokkaido, and a comparison between genetic diversity based on predefined populations and inferred genetic populations was conducted.
Methods
Study area and sample collection
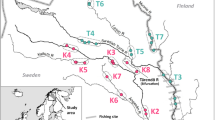
In 2013 and 2014, a total of 928 juvenile masu salmon were collected from 16 rivers in Hokkaido, Japan, by electrofishing (Table 1 and Fig. 1, Supplementary Table S1). Most of these rivers are small- to medium-sizes rivers (13–50 km in length), although two rivers (GOK and OSA) were very small (ca. < 7 km, Supplementary Table S1). In these rivers, there was no official record of artificial releases, thus the samples used in this study were thought to be native fish. Juveniles were collected randomly with regard to age structure. As a result, juveniles included young-of-the-year (i.e. age-0) and 1-year-old fish, although the majority were age-0 fish. Adipose fines or caudal fins were preserved in 99.5% ethanol until DNA extraction.

Map of the sampling sites of 16 wild masu salmon populations in Hokkaido, Japan
Microsatellite DNA experiment
Genomic DNA was extracted using Chelex 100 (Bio-Rad, CA, USA) according to the manufacturer’s instructions. Fifteen microsatellite loci developed from other salmonids (One110, One111, Olsen et al. 2000; OMM1140, Rexroad et al. 2001; OMM1148, Rexroad et al. 2002; OMM1316, OMM1325, OMM1344, OMM1347, Palti et al. 2002; OMM1352, OMM1358, Palti et al. 2003; OMM1375, OMM1402, OMM1404, Rodriguez et al. 2003; Oma3ke, Oma4my, Noguchi et al. 2003) were amplified in an automated thermal cycler (PCR thermal cycler SP, TAKARA, Otsu, Japan). Polymerase chain reaction (PCR) amplifications and procedures of genotyping were performed in accordance with the procedures described by Kitanishi et al. (2017a). PCR products were analyzed using an ABI PRISM 3730 Genetic Analyzer and Genemapper ver. 4.0 (Applied Biosystems, Foster City, CA, USA).
Data analyses
To examine the deviation from the Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium (LD), the exact HWE test and composite LD test, as implemented in GENEPOP 3.4 (Raymond and Rousset 1995) were used, respectively. Significance levels for the HWE and LD tests were assessed following the false discovery rate (FDR, Benjamini and Yekutieli 2001). Expected heterozygosity (HE) and allelic richness (Ar, El Mousadik and Petit 1996) were calculated using FSTAT 2.9.3 (Goudet 2001). The effective number of breeders (Nb) of each sample was estimated by applying LD method (Waples and Do 2008). In this procedure, a contemporary estimator of the effective number of populations (Ne_LDNE) was estimated at first using LDNE (Waples and Do 2008). Following Waples and Do (2008), a value of 0.02 as the lowest allele frequency and the jackknife method to measure the 95% confidence interval (CI) were used. Then, the estimated values of Ne_LDNE were adjusted using the method developed by Waples et al. (2014) (Nb(adj2), see Table 3 in Waples et al. 2014) that corrects bias due to overlapping generations using two life history traits, that is adult life span (AL) and age at maturity (α). In this study, AL = 4 and α = 1 were used (Sugiwaka and Kojima 1984; Kato 1991). Finally, the program BOTTLENECK (Piry et al. 1999) was used to test for recent bottlenecks. The two-phase model of mutation with 95% stepwise mutation model and a 12% variance was applied, as recommended for microsatellite (Piry et al. 1999). Statistical significance was tested with a Wilcoxon signed rank test and 1000 iterations.
Population structure
To infer spatial genetic structuring and assign individuals to K groups in Hokkaido, we conducted a Bayesian assignment test implemented in the STRUCTURE software (Pritchard et al. 2000). This analysis was conducted hierarchically, and the first analysis was conducted using the entire dataset of 16 samples to infer the number of genetic groups in Hokkaido. Then, a second analyses using as input the groups identified by the first analysis was conducted. In the STRUCTURE analyses, the admixture model was used, and 10 independent runs were performed for each value of K. The number of iterations was 500,000 after a burn-in period of 200,000. STRUCTURE outputs were submitted to STRUCTURE HARVESTER (Earl and Vonholdt 2012) to determine the most likely value of K using ∆k method (Evanno et al. 2005).
For the analysis of population genetic structure, an analysis of molecular variance (AMOVA) was conducted using Arlequin 3.5 (Excoffier et al. 2005) for the three genetic groups identified by the STRUCTURE analysis (Analysis 1, see Results) and three geographic groups based on geographic locations (i.e. the Pacific, Sea of Okhotsk, and Sea of Japan groups; Analysis 2). The extent of genetic differentiation among the samples was evaluated by FST using GenAlEx 6.5 (Peakall and Smouse 2006), and the significance levels of these tests were corrected following the FDR. To visualize the genetic relationship of the 16 masu salmon samples, a principal coordinate analysis (PCoA) was conducted using the matrix of pairwise FST using GenAlEx. Isolation by distance (IBD) was examined by regressing genetic differentiation on geographic distance (km). In this analysis, FST/(1-FST) and coastal distance were used as the genetic and geographic distances, respectively, and correlations between genetic and geographic distance were evaluated using IBDWS ver. 3.23 (Jensen et al. 2005).
Decomposed pairwise regression (DPR, Koizumi et al. 2006) was conducted to detect outlier populations. Briefly, the populations with the highest or lowest mean residuals in the regression of pairwise genetic and geographic distances were sequentially removed from the analysis if the 95% confidence interval of the mean residual value did not include 0 (process 1, which identified “putative” outliers). The putative outlier populations were then identified by choosing the best model with and without outliers based on Akaike information criteria (AIC) value (process 2, which identified “true” outliers), where a model with a smaller AIC was plausible. Then, for each population, pairwise genetic and geographic distances were regressed separately including outlier populations against the non-outlier populations (Koizumi et al. 2006). This analysis was also conducted hierarchically, and the first analysis was conducted using the entire dataset of 16 samples to infer outlier populations. Then, we conducted a second DPR analyses using as input the groups identified by STRUCTURE analysis.
Results
Sample statistics
The exact test of HWE revealed a significant global trend toward a heterozygote deficiency (p < 0.001). One sample (i.e., OSA) exhibited a significant deviation following FDR, and five out of 240 cases deviated significantly. This trend toward heterozygote deficiency was not caused by one particular locus because Kendall’s test of concordance showed that the ranks of all loci, as estimated by FIS, were not consistent across samples (Kendall’s W = 0.043, p > 0.05). No case of LD was detected systematically for the same pair of loci across different populations. The mean number of alleles, allelic richness (Ar), and number of private alleles within populations ranged from 4.7 to 8.9, from 4.5 to 8.3, and from 0 to 6, respectively (Table 1). Mean expected heterozygosity (HE) was similar for each sample, varying from 0.61 to 0.67, except for OSA and ONB (Table 1). The effective number of breeders (Nb(adj2)) for each sample ranged from 11.5 (9.5–13.9) to 831.2 (298.3–∞) and, with the some exceptions, many of the estimated Nb(adj2) were relatively small (ca. Nb(adj2) < 150, Table 1). Program BOTTLENECK showed that there was no evidence of recent bottleneck.
Genetic structure
The initial STRUCTURE analysis indicated K = 3 and suggested the existence of two genetically distinct groups that were strongly associated with geographical regions: northeastern and southwestern regions (Fig. 1 and 2a); and one group with the only population (OSA). A second STRUCTURE analyses was conducted for northeastern and southwestern groups. For northeastern group, the second round of STRUCTURE analysis assigned individuals to six groups (K = 6, Fig. 2b). Two populations located in the northern part of the Sea of Okhotsk (SNA and OUM) constituted the same group, whereas the others were different from one another. For the southwestern group, five genetic groups (K = 5) were identified and, except for the populations located in mid-western Hokkaido (i.e. GOK and ATS), genetic similarity did not correspond to their geographic proximities (Fig. 2c).

Estimated population clustering inferred by two levels of hierarchical STRUCTURE analysis of 16 wild masu salmon populations (a). Horizontal line represents each individual, and different color represent independent cluster. Black lines separate individuals from different populations. Arrows delineate the progress of the hierarchical approach, where subsets of the data were subsequently analyzed, and colors of each arrow correspond to the color of each cluster in the 1st level the analysis (i.e. red: northeastern group (b) green: southwestern group (c))
AMOVA analyses were conducted based on the STRUCTURE results (Analysis 1) and on the geographic locations of each sample (Analysis 2; Pacific, Sea of Okhotsk, and Sea of Japan). For the Analysis 1, AMOVA revealed that 2.47% of the total genetic variation was partitioned into among three groups (p < 0.001, Table 2) and 3.77% was explained by differences among samples within group level (p < 0.001). For Analysis 2, genetic variation at among groups level was much lower (0.05% of total genetic variation, p > 0.05) than that of Analysis 1.
For pairwise comparisons, significant genetic differentiation was observed among all 16 populations after FDR (range 0.0112 to 0.1062, Supplementary Table S2), with a global FST of 0.0581 (p < 0.001). The global FST of the northeastern and southwestern groups identified by the STRUCTURE analysis were 0.043 and 0.040, respectively (p < 0.001 in both cases). One population (OSA) showed remarkable genetic differentiations from all other populations (range 0.0515 to 0.1062). PCoA plots illustrated that three populations (i.e. OSA, ONB, and NUK) were distant from other populations (Fig. 3). The remaining populations in the center, except for GOK, were clustered reflecting their genetic groups. There was no significant IBD for the 16 populations (r = 0.056, p = 0.106, Fig. 4). However, the Mantel test revealed significant IBD for the northeastern group (r = 0.366, p = 0.049), but did not for the southwestern group (r = −0.050, p = 0.623).

Plots of the principle coordinate analysis. Filled and grey circles represent plots of northeastern and southwestern groups identified by STRUCTURE analysis, respectively. For abbreviations, see Table 1

Relationship between genetic and geographic distance. Filled and grey circles represent plots of northeastern and southwestern groups identified by STRUCTURE analysis, respectively. Open circles represent plots of between groups
For the first DPR analysis, based on the bias of the regression residuals (process 1), 10 populations were identified as putative outlier populations, and these populations, except for two populations (UTS and ONB), were located in from the middle to southern part of Hokkaido (OSA, NUK, SHA, KER, NOD, SEK, HOR, and ATS). However, based on the AIC values (process 2), no population was identified as a true outlier (data not shown). A second DPR analyses for southwestern group, although five populations (NUK, SHA, GOK, ATS, and HOR) were identified as putative outlier populations, there was no outlier population based on the AIC values. Similarly, for northeastern group, no populations was identified as a true outlier population although two populations (ONB and UTS) were identified as putative outliers.
Discussion
This study found that the native masu salmon populations in Hokkaido were divided into two genetically distinct groups. The northeastern group was primarily composed on populations along the Sea of Okhotsk, the Nemuro Strait, and eastern Pacific from Cape Erimo and the southwestern group was primarily composed of the populations along the Sea of Japan and western part of Pacific coast from Cape Erimo. Although this result supported previous studies that reported the existence of regional genetic groups in Hokkaido (Okazaki 1986; Suzuki et al. 2000; Kitanishi et al. 2007; Yu et al. 2010), the boundary observed in this study is different from these studies and is in agreement with differences in life history traits of masu salmon. It has been reported that life history traits differ between populations inhabiting northeastern Hokkaido and those in the southwestern region. For instance, the spawning season of masu salmon in northeastern Hokkaido (the Sea of Okhotsk and the Nemuro Strait coast) is approximately 2–4 weeks earlier than in the southwestern region (southern part of the Sea of Japan coast) (Kato 1991), and this difference may facilitate divergence between these two regions. In particular, difference in timing of spawning could have a large effect on genetic structure because it may lead to genetic divergence (i.e., isolation by time, reviewed by Hendry and Day 2005). The fact that other life-history traits and morphological characteristics, such as body size, fecundity, and sex ratio of anadromous individuals, differ between northeastern and southwestern Hokkaido (Kato 1991) also supports the existence of the observed population boundary. In addition, differences in abiotic factors, such as geographic features and water temperature in each river may also influence population genetic structure. Therefore, the observed boundary could be attributed to the differences in life history traits as well as abiotic factors.
Although there are two major genetic groups in Hokkaido, these two groups may exhibit different genetic structures. In the northeastern group, a significant IBD was observed, whereas in the southeastern group, IBD was not detected. The difference between the northeastern and southwestern groups could be attributed to the difference in historical processes. During the glacial period, masu salmon populations in northern and eastern Hokkaido were thought to have undergone severe depopulation (Kitanishi et al. 2007), similar to other salmonid fish in the Far East (Brykov et al. 2003). Following deglaciation, populations in northeastern Hokkaido were constructed by the northward recolonization from refugia (see below) along the currents. This migration pattern (i.e., one-dimensional dispersal, Slatkin and Maddison 1990) and a shorter population history (Crispo and Hendry 2005) might have enhanced the IBD in the northeastern populations. In contrast, southwestern Hokkaido may serve as refugia and masu salmon populations in this area may be relatively stable (Kitanishi et al. 2007; Yu et al. 2010). However, it was reported that among-river dispersal of masu salmon was much less common (Kitanishi et al. 2017c), and the ratio of anadromous fish decreases with decreasing latitude (Kato 1991). These facts may simply reduce among-river dispersal, especially in southern regions, and populations in southwestern Hokkaido may remain independent for a long period (Castric and Bernatchez 2003). In fact, the DPR analysis implied larger genetic divergences among the southwestern populations. The fact that the boundary between the northeastern and southwestern groups is consistent with the northern limit of glacial refugia (i.e. 55 °N; Kitanishi et al. 2007) supports the effects of glaciation on genetic structure. Therefore, a different population history, as well as dispersal route (Castric and Bernatchez 2003) and time since colonization (Crispo and Hendry 2005), influenced by glaciation could have resulted in different genetic structure for the northeastern and southwestern groups of masu salmon.
Regarding within-group genetic structure, significant genetic differentiations estimated by FST were observed among all population pairs. In addition, STRUCTURE analysis implied that all populations, except for geographically proximate populations (e.g. GOK and ATS), were different from one another. Moreover, all populations, except for ATS, possessed a number of private alleles. These results suggest that masu salmon populations are relatively independent from each other because of precise homing. Previous studies using several genetic markers reported relatively high genetic differentiation and suggested that genetic differentiation could be attributed to precise homing (e.g., Suzuki et al. 2000; Kitanishi et al. 2007). Precise homing of masu salmon has been reported by previous studies using physical markings (Mayama et al. 1988; Miyakoshi et al. 2012) and genetic markers (Kitanishi et al. 2017c). Such precise homing could lead to genetic divergence and/or genetic structuring even at small spatial scales. In addition, exclusion of artificial releases may also reinforce genetic dissimilarity of each population (Eldridge and Naish 2007). Therefore, masu salmon populations in Hokkaido are different from each other due to precise homing, despite being roughly divided into two genetically distinct groups.
Nevertheless, genetic characteristics of some populations (OSA, ONB, and NUK) were substantially different from the other populations. Among these, OSA exhibited remarkable genetic divergence from both the northeastern and southwestern groups. This divergence may be caused by genetic drift and/or a founder effect because of small population size. This river is very small (ca. 7 km in length, 2–3 m in width, Supporting Table S1) and the waterway may sometimes be undeground, similar to it’s neighboring river. In such a small population, genetic composition may fluctuate or change from year to year (Jorde and Ryman 1996; Østergaard et al. 2003; Heggenes et al. 2009). Another possibility that could lead to the greater genetic divergence is that masu salmon inhabiting OSA have a unique genetic composition, although the causative factors are unknown. To identify the effects of genetic drift, uniqueness, and their combination on the greater genetic divergence in OSA, further studies using samples collected over several years would be necessary. In contrast to OSA, ONB and NUK are approximately 43 and 33 km long, respectively (Supporting Table S1), and consequently the processes proposed of the greater genetic differentiation in OSA are not applicable. However, small population size, possibly caused by set-net fishing, was observed in ONB (Kitanishi unpubl. data), and releases of hatchery-produced masu salmon by private entities were sometime conducted in NUK, although the number of released fish was relatively small (2000–54,000/year, Salmon and Freshwater Fisheries Research Institute, Hokkaido Research Organization). Therefore, the larger genetic differentiation observed in ONB and NUK may be due to founder effects and genetic introgression that could be caused by anthropogenic activities.
Finally, the results of this study highlight the importance of inferred populations based on their genetic features rather than predefined populations based on geographic locations to infer correct genetic structure. Generally, predefined populations based on geographical characteristics, such as capes and coastal regions, have been a standard approach in empirical genetic studies. In Japan, such a predefined approach has been used in previous salmonid studies including masu and chum salmon (i.e., Pacific, Sea of Okhotsk, and Sea of Japan, Kitanishi et al. 2007; Yu et al. 2012). However, this study revealed that the boundary between the genetic groups did not correspond to geographical location. The AMOVA suggested that the predefined approach could have possibly led to a misinterpretation of genetic structuring. Defining a population boundary and characterization of indigenous genetic features are first step for planning effective conservation or management strategies (Moritz 1994; Frankham et al. 2004), however this study cautions that genetic grouping could easily be affected by the defining populations. Therefore, sampling and analysis design of samples for inferring spatial genetic structuring should be mindful of their potential influences.
Although there was no signals of recent bottleneck in the analyzed populations, the effective number of breeders were relatively low and, in fact, masu salmon resources is decreasing in Hokkaido (Salmon and Freshwater Fisheries Research Institute, Hokkaido Research Organization). This study showed that each river population could have indigenous genetic characteristics primarily, and temporal changes of genetic structure was also reported in masu salmon (Kitanishi et al. 2017a). Given these findings, the possibility exists that genetic diversity and indigenous genetic features within local populations become decreasingly. Therefore, treatment of each regional population as separate management unit and periodic monitoring over time and space are necessary to ensure the persistence of the genetic diversity and indigenous genetic features that adapt for local habitat. In masu salmon, adaptive management approaches to balance conservation of native masu salmon resources with resource enhancement are being promoted (Nagata et al. 2012a, b), and the results of this study provide a genetic basis and could contribute to determine prioritizing areas for conservation of native masu salmon as well as to improve the effectiveness of the management program. For instance, our finding, the two major genetic groups exist in Hokkaido, indicates that optimal management strategies would be different between the groups (regions). In particular, treatment of each population as a separate management unit would be important for the southwestern group due to the reduction of among-river connectivities (see above paragraph). Similarly, reduction of local populations or subspecies, loss of genetic diversity, genetic homogenization due to artificial releases of non-natives, and population definitions (see above paragraph), are also true for species whose movement is restricted, such as freshwater fish (e.g., Mabuchi et al. 2008; Watanabe 2012; Faulks et al. 2017). Thus, accumulation of the knowledge of detailed genetic structuring would allow for the development of personalized conservation and management strategies that adapt for the specific characteristics of each population or management unit.
References
Allendorf FW, Leary RF, Spruell P, Wenburg JK (2001) The problems with hybrids: setting conservation guidelines. Trends Ecol Evol 16:613–622
Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29:1165–1188
Brykov VA, Polyakova NE, Prokhorova AV (2003) Phylogeographic analysis of chum salmon Oncorhynchus keta walbaum in Asian populations based on mtDNA variation. Russ J Genet 39(1):61–67. https://doi.org/10.1023/A:1022074911828
Caputo V, Giovannotti M, Cerioni PN, Caniglia ML, Splendiani A (2004) Genetic diversity of brown trout in central Italy. J Fish Biol 65(2):403–418. https://doi.org/10.1111/j.0022-1112.2004.00458.x
Castric V, Bernatchez L (2003) The rise and fall of isolation by distance in the anadromous brook charr (Salvelinus fontinalis Mitchill). Genetics 163(3):983–996
Corander J, Waldmann P, Marttinen P, Sillanpää MJ (2004) BAPS 2: enhanced possibilities for the analysis of genetic population structure. Bioinformatics 20(15):2363–2369. https://doi.org/10.1093/bioinformatics/bth250
Crispo E, Hendry AP (2005) Does time since colonization influence isolation by distance? A meta-analysis. Conserv Genet 6(5):665–682. https://doi.org/10.1007/s10592-005-9026-4
Earl DA, Vonholdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361. https://doi.org/10.1007/s12686-011-9548-7
Edpalina RR, Yoon M, Urawa S, Kusuda S, Urano A, Abe S (2004) Genetic variation in wild and hatchery populations of masu salmon (Oncorhynchus masou) inferred from mitochondrial DNA sequence analysis. Fish Genet Breed Sci 34:37–44
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Genet 92(7):832–839. https://doi.org/10.1007/BF00221895
Eldridge WH, Naish KA (2007) Long-term effects of translocation and release numbers of fine-scale population structure among coho salmon (Oncorhynchus kisutch). Mol Ecol 16(12):2407–2421. https://doi.org/10.1111/j.1365-294X.2007.03271.x
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinforma 1:47–50
Faulks LK, Kerezsy A, Unmack PJ, Johnson JB, Hughes JM (2017) Going, going, gone? Loss of genetic diversity in two critically endangered Australian freshwater fishes, Scaturiginichthys vermeilipinnis and Chlamydogobius squamigenus, from Great Artesian Basin springs at Edgbaston, Queensland, Australia. Aquat Conserv 27:39–50
Frankham R, Ballow JD, Briscoe DA (2004) A primer of conservation genetics. Cambridge University Press, Cambridge. https://doi.org/10.1017/CBO9780511817359
Fukushima M, Kameyama S, Kaneko M, Nakao K, Steel EA (2007) Modelling the effects of dams on freshwater fish distributions in Hokkaido, Japan. Freshw Biol 52(8):1511–1524. https://doi.org/10.1111/J.1365-2427.2007.01783.X
Glover KA, Quintela M, Wennevik V, Besnier F, Sorvik AGE, Skaala O (2012) Three decades of farmed escapees in the wild: a spatio-temporal analysis of Atlantic salmon population genetic structure throughout Norway. PLoS One 7(8):e43129. https://doi.org/10.1371/journal.pone.0043129
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3) Available from: http://www2.unil.ch/popgen/softwares/fstat.htm
Heggenes J, Røed KH, Jorde PE, Brabrand A (2009) Dynamic micro-geographic and temporal genetic diversity in vertebrates: the case of lake-spawning populations of brown trout (Salmo trutta). Mol Ecol 18(6):1100–1111. https://doi.org/10.1111/j.1365-294X.2009.04101.x
Hendry AP, Day T (2005) Population structure attributable to reproductive time: isolation by time and adaptation by time. Mol Ecol 14(4):901–916. https://doi.org/10.1111/j.1365-294X.2005.02480.x
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet 6(1):13. https://doi.org/10.1186/1471-2156-6-13
Jorde PE, Ryman N (1996) Demographic genetics of brown trout (Salmo trutta) and estimation of effective population size from temporal change of allele frequencies. Genetics 143(3):1369–1381
Kato F (1991) Life histories of masu and amago salmon (Oncorhynchus masou and Oncorhynchus rhodurus). In: Groot C, Margolis L (eds) Pacific Salmon life histories. UBC Press, Vancouver, pp 447–520
Kawamura K, Kubota M, Furukawa M, Harada Y (2007) The genetic structure of endangered indigenous populations of the amago salmon, Oncorhynchus masou ishikawae, in Japan. Conserv Genet 8(5):1163–1176. https://doi.org/10.1007/s10592-006-9271-1
Kitanishi S, Edo K, Yamamoto T, Azuma N, Hasegawa O, Higashi S (2007) Genetic structure of masu salmon (Oncorhynchus masou) populations in Hokkaido, northernmost Japan, inferred from mitochondrial DNA variation. J Fish Biol 71(sc):437–452. https://doi.org/10.1111/J.1095-8649.2007.01689.X
Kitanishi S, Yamamoto T, Higashi S (2009) Microsatellite variation reveals fine-scale genetic structure of masu salmon, Oncorhynchus masou, within the Atsuta River. Ecol Freshw Fish 18(1):65–71. https://doi.org/10.1111/J.1600-0633.2008.00325.X
Kitanishi S, Ikeda T, Yamamoto T (2017a) Short-term temporal instability in fine-scale genetic structure of masu salmon. Freshw Biol 62(9):1655–1664. https://doi.org/10.1111/fwb.12978
Kitanishi S, Mukai T, Yamamoto T, Tago Y, Oda M (2017b) Genetic introgression between masu salmon Oncorhynchus masou masou and amago salmon Oncorhynchus masou ishikawae. Nippon Suisan Gakkaishi 83(3):400–402. (in Japanese with English abstract). https://doi.org/10.2331/suisan.16-00082
Kitanishi S, Yamamoto T, Ishii H, Yamaguchi Y, Kobayashi T (2017c) Dispersal patterns of anadromous and freshwater resident masu salmon at different spatial scales in mid-western Hokkaido, Japan. Ichthyol Res 64(1):111–115. https://doi.org/10.1007/s10228-016-0525-8
Koizumi I, Yamamoto S, Maekawa K (2006) Decomposed pairwise regression analysis of genetic and geographic distances reveals a metapopulation structure of stream-dwelling Dolly Varden charr. Mol Ecol 15(11):3175–3189. https://doi.org/10.1111/j.1365-294X.2006.03019.x
Mabuchi K, Senou H, Nishida M (2008) Mitochondrial DNA analysis reveals cryptic large-scale invasion of non-native genotypes of common carp (Cyprinus carpio) in Japan. Mol Ecol 17:796–809
Mayama H, Nomura T, Ohkuma K (1988) Seaward migration and adult return of the marked masu salmon, Oncorhynchus masou, released in late fall before wintering. Sci Rep Hokkaido Salm Hatch 42:21–36
Miyakoshi Y, Nagata M, Sugiwaka K, Kitada S (2004) Evaluation of stock enhancement programmes for masu salmon in Hokkaido, northern Japan, by two-stage sampling surveys of commercial landings. In: Leber KM, Kitada S, Blankenship HL, Svåsand T (eds) Stock enhancement and sea ranching, 2nd edn. Blackwell, Oxford. https://doi.org/10.1002/9780470751329.ch16
Miyakoshi Y, Takahashi M, Ohkuma K, Urabe H, Shimoda K, Kawamula H (2012) Homing of masu salmon in the tributaries of the Shiribetsu River evaluated by returns of marked fish. Sci Rep Hokkaido Fish Res Inst 81:125–129
Morita K, Saito T, Miyakoshi Y, Fukuwaka M, Nagasawa T, Kaeriyama M (2006) A review of Pacific salmon hatchery programmes on Hokkaido Island, Japan. ICES J Mar Sci 63(7):1353–1363. https://doi.org/10.1016/j.icesjms.2006.03.024
Moritz C (1994) Defining 'evolutinarily significant units' for conservation. Trends Ecol Evol 9:373–375
Nagata M, Miyakoshi Y, Urabe H, Fujiwara M, Sasaki Y, Kasugai K, Torao M, Ando D, Kaeriyama M (2012a) Erratum to: an overview of salmon enhancement and the need to manage and monitor natural spawning in Hokkaido, Japan. Environ Biol Fish 94(1):359–361. https://doi.org/10.1007/s10641-011-9945-5
Nagata M, Miyakoshi Y, Urabe H, Fujiwara M, Sasaki Y, Kasugai K, Torao M, Ando D, Kaeriyama M (2012b) An overview of salmon enhancement and the need to manage and monitor natural spawning in Hokkaido, Japan. Environ Biol Fish 94(1):311–323. https://doi.org/10.1007/s10641-011-9882-3
Noguchi D, Ikeda M, Nakajima M, Taniguchi N (2003) Isolation and characterization of microsatellite DNA markers for population genetics study of masu salmon, Oncorhynchus masou masou. Fish Genet Breed Sci 33:61–66
Okazaki T (1986) Genetic variation and population structure in masu salmon Oncorhynchus masou of Japan. Bull Jpn Soc Sci Fish 52(8):1365–1376. https://doi.org/10.2331/suisan.52.1365
Olsen JB, Wilson SL, Kretschmer EJ, Jones KC, Seeb JE (2000) Characterization of 14 tetranucleotide microsatellite loci derived from sockeye salmon. Mol Ecol 9(12):2185–2187. https://doi.org/10.1046/j.1365-294X.2000.105317.x
Østergaard S, Hansen MM, Loeschcke V, Nielsen EE (2003) Long-term temporal changes of genetic composition in brown trout (Salmo trutta L.) populations inhabiting an unstable environment. Mol Ecol 12(11):3123–3135
Palsbøll PJ, Berube M, Allendorf FW (2007) Identification of management units using population genetic data. Trends Ecol Evol 22(1):11–16. https://doi.org/10.1016/j.tree.2006.09.003
Palti Y, Fincham MR, Rexroad CE (2002) Characterization of 38 polymorphic microsatellite markers for rainbow trout (Oncorhynchus mykiss). Mol Ecol Notes 2(4):449–452. https://doi.org/10.1046/j.1471-8278.2002.00274.x
Palti Y, Danzmann RG, Rexroad CE (2003) Characterization and mapping of 19 polymorphic microsatellite markers for rainbow trout (Oncorhynchus mykiss). Anim Genet 34(2):153–156. https://doi.org/10.1046/j.1365-2052.2003.00965_7.x
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Notes 6(1):288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Raymond M, Rousset F (1995) Genepop (Version-1.2): population genetics software for exact tests and ecumenicism. J Hered 86(3):248–249. https://doi.org/10.1093/oxfordjournals.jhered.a111573
Rexroad CE, Coleman RL, Martin AM, Hershberger WK, Killefer J (2001) Thirty-five polymorphic microsatellite markers for rainbow trout (Oncorhynchus mykiss). Anim Genet 32(5):317–319. https://doi.org/10.1046/j.1365-2052.2001.0730b.x
Rexroad CE, Coleman RL, Hershberger WK, Killefer J (2002) Eighteen polymorphic microsatellite markers for rainbow trout (Oncorhynchus mykiss). Anim Genet 33(1):76–78. https://doi.org/10.1046/j.1365-2052.2002.0742d.x
Rodriguez F, Rexroad CE, Palti Y (2003) Characterization of twenty-four microsatellite markers for rainbow trout (Oncorhynchus mykiss). Mol Ecol Notes 3(4):619–622. https://doi.org/10.1046/j.1471-8286.2003.00531.x
Slatkin M, Maddison WP (1990) Detecting isolation by distance using phylogenies of genes. Genetics 126(1):249–260
Sugiwaka K-i, Kojima H (1984) Influence of individual density on smoltification in wild juvenile masu salmon (Oncorhynchus masou) in the Atsuta River. Sci Rep Hokkaido Fish Hatchery 39:19–37
Suzuki K-iT, Kobayashi T, Matsuishi T, K-i N (2000) Genetic variability of masu salmon in Hokkaido, by restriction fragment length polymorphism analysis of mitochondrial DNA. Nippon Suisan Gakkaishi 66(4):639–646. https://doi.org/10.2331/suisan.66.639
Tamate T, Hayajiri M (2008) The relationship between the number of main dams and the coastal catch of masu salmon (Oncorhynchus masou) in Hokkaido: implications for river ecosystem conservation. Water Sci 52:72–84
Vähä JP, Erkinaro J, Niemela E, Primmer CR (2007) Life-history and habitat features influence the within-river genetic structure of Atlantic salmon. Mol Ecol 16(13):2638–2654. https://doi.org/10.1111/j.1365-294X.2007.03329.x
Waples RS, Do C (2008) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8(4):753–756. https://doi.org/10.1111/j.1755-0998.2007.02061.x
Waples RS, Gaggiotti O (2006) What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol 15(6):1419–1439. https://doi.org/10.1111/j.1365-294X.2006.02890.x
Waples RS, Antao T, Luikart G (2014) Effects of overlapping generations on linkage disequilibrium estimates of effective population size. Genetics 197(2):769–780. https://doi.org/10.1534/genetics.114.164822
Watanabe K (2012) Faunal structure of Japanese freshwater fishes and its artificial disturbance. Environ Biol Fish 94(3):533–547. https://doi.org/10.1007/s10641-010-9601-5
Yamamoto T, Reinhardt UG (2003) Dominance and predator avoidance in domesticated and wild masu salmon Oncorhynchus masou. Fish Sci 69(1):88–94. https://doi.org/10.1046/j.1444-2906.2003.00591.x
Yamamoto S, Morita K, Koizumi I, Maekawa K (2004) Genetic differentiation of white-spotted charr (Salvelinus leucomaenis) populations after habitat fragmentation: spatial-temporal changes in gene frequencies. Conserv Genet 5(4):529–538. https://doi.org/10.1023/B:COGE.0000041029.38961.a0
Yu J-N, Azuma N, Yoon M, Brykov V, Urawa S, Nagata M, Jin DH, Abe S (2010) Population genetic structure and Phylogeography of Masu Salmon (Oncorhynchus masou masou) inferred from mitochondrial and microsatellite DNA analyses. Zool Sci 27(5):375–385. https://doi.org/10.2108/Zsj.27.375
Yu J-N, Azuma N, Abe S (2012) Genetic differentiation between collections of hatchery and wild masu salmon (Oncorhynchus masou) inferred from mitochondrial and microsatellite DNA analyses. Environ Biol Fish 94(1):259–271. https://doi.org/10.1007/s10641-011-9869-0
Acknowledgements
The authors are grateful to members of Salmon and Freshwater Fisheries Research Institute, Hokkaido Research Organization and members of Hokkaido Salmon Propagation Association for their helping in collecting samples. We are grateful to two anonymous reviewers for their constructive comments on this manuscript. This study was conducted with the permission of the Hokkaido Government.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kitanishi, S., Yamamoto, T., Urabe, H. et al. Hierarchical genetic structure of native masu salmon populations in Hokkaido, Japan. Environ Biol Fish 101, 699–710 (2018). https://doi.org/10.1007/s10641-018-0730-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10641-018-0730-6