
Summary
D2C7-(scdsFv)-PE38KDEL (D2C7-IT) is a novel immunotoxin that reacts with wild-type epidermal growth factor receptor (EGFRwt) and mutant EGFRvIII proteins overexpressed in glioblastomas. This study assessed the toxicity of intracerebral administration of D2C7-IT to support an initial Food and Drug Administration Investigational New Drug application. After the optimization of the formulation and administration, two cohorts (an acute and chronic cohort necropsied on study days 5 and 34) of Sprague–Dawley (SD) rats (four groups of 5 males and 5 females) were infused with the D2C7-IT formulation at total doses of 0, 0.05, 0.1, 0.4 μg (the acute cohort) and 0, 0.05, 0.1, 0.35 μg (the chronic cohort) for approximately 72 h by intracerebral convection-enhanced delivery using osmotic pumps. Mortality was observed in the 0.40 μg (5/10 rats) and 0.35 μg (4/10 rats) high-dose groups of each cohort. Body weight loss and abnormal behavior were only revealed in the rats treated with high doses of D2C7-IT. No dose-related effects were observed in clinical laboratory tests in either cohort. A gross pathologic examination of systemic tissues from the high-dose and control groups in both cohorts exhibited no dose-related or drug-related pathologic findings. Brain histopathology revealed the frequent occurrence of dose-related encephalomalacia, edema, and demyelination in the high-dose groups of both cohorts. In this study, the maximum tolerated dose of D2C7-IT was determined to be between 0.10 and 0.35 μg, and the no-observed-adverse-effect-level was 0.05 μg in SD rats. Both parameters were utilized to design the Phase I/II D2C7-IT clinical trial.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glioblastoma is the most aggressive malignant brain tumor among all primary brain and central nervous system (CNS) tumors diagnosed in the United States. Glioblastomas represented 45.6 % of all malignant brain tumors from 2007 to 2011, according to a statistical report from the Central Brain Tumor Registry of the United States (CBTRUS) [1]. The median survival time for glioblastoma patients given the standard treatment of surgery followed by radiation and chemotherapy is less than 15 months [1, 2]. Even with newly developed agents, such as temozolomide or bevacizumab, there is only a slight improvement in the survival time of glioblastoma patients [2]. Thus, there is an urgent need for more advanced and efficient therapeutic approaches to improve the poor survival outlook of glioblastoma patients.
Recombinant immunotoxins (ITs) may provide a much-needed breakthrough in glioblastoma therapy. In the past two decades, monoclonal antibody (mAb)-based studies have increasingly focused on ITs that are constructed by fusing genetically engineered single-chain variable region antibody fragments (scFv), which target antigens specifically expressed by brain tumor cells, to bacterial or plant toxins. Because the scFv-IT fusion protein is smaller than the original immunoglobulin (IgG), it has a superior capacity for tumor penetration, which can lead to enhanced anti-tumor therapeutic efficacy when it is delivered intrathecally, intratumorally, or intracranially [3–7].
Previous studies have reported the amplification of the epidermal growth factor receptor (EGFR) in 40 % to 50 % of glioblastoma patients [6]. In contrast, the EGFR level in the normal brain is undetectable or extremely low [8]. In correlation with EGFR gene amplification, the protein is overexpressed in approximately 60 % to 90 % of glioblastoma patients. Even in the absence of gene amplification, 12 % to 38 % of glioblastoma patients exhibit EGFR protein overexpression, which may be due to aberrant translational and post-translational mechanisms [9]. Moreover, up to 67 % of all tumors with wild-type EGFR (EGFRwt) amplification express a truncated variant of EGFR, known as EGFR variant III (EGFRvIII), which lacks the extracellular ligand-binding domain (exons 2 through 7) [2, 10]. The development of mAb-based agents that can target both forms of the receptor for therapy would be advantageous because of the high prevalence of the EGFRvIII mutation in glioblastomas with EGFRwt amplification.
We have developed a novel targeted IT, D2C7-(scdsFv)-PE38KDEL (D2C7-IT), which reacts with both the EGFRwt and EGFRvIII proteins by fusing the scFv of the D2C7 mAb with domains II and III of Pseudomonas exotoxin A (PE38KDEL) [11]. In vitro cytotoxicity data shows that D2C7-IT effectively inhibited protein synthesis in a variety of EGFRwt-, EGFRvIII-, or both EGFRwt- and EGFRvIII-expressing glioblastoma xenograft cells and human tumor cell lines. Notably, in intracranial animal models of EGFRwt-expressing and EGFRwt-EGFRvIII-expressing glioblastoma xenografts, intracranial administration of D2C7-IT via convection-enhanced delivery (CED) has been shown to increase survival by more than 150 % [11, 12]. Hence, D2C7-IT should be clinically efficacious against brain tumors expressing either EGFRwt or EGFRvIII, or expressing both EGFRwt and EGFRvIII. A preclinical study was performed under Good Laboratory Practice (GLP) regulations to evaluate the systemic toxicity of D2C7-IT administered via intracerebral CED using an osmotic pump in rats to support an initial US Food and Drug Administration (FDA) Investigational New Drug (IND) application for a Phase I/II clinical trial in patients with glioblastoma [12, 13]. In patients, the distribution of IT by intracerebral CED can be monitored by co-infusion with a low-molecular-weight tracer, gadolinium-diethylene triamine pentaacetic acid (Gd-DTPA), and 124I-labeled human serum albumin (124I-HSA) [14]. The IT was co-infused with Gd-DTPA and 124I-HSA in this current rat GLP study with the aim of replicating this formulation in the future D2C7-IT clinical trial.
Materials and methods
Animal care
Equal numbers of adult male and female Sprague–Dawley (SD) rats were selected for use in this study, and all rats were received from Charles River Laboratories, Inc. (Portage, MI, USA). Upon arrival, the rats were visually inspected for general health and acceptability for use in this study, and the numbers of rats and sex were verified. Each rat received a unique identification number following randomization into the different groups. The rats were approximately 13 to 17 weeks of age at the initiation of the study. At the beginning of treatment, the body weights of the rats were 360–460 g for the males and 210–290 g for the females. The animal housing and care, study protocol, and standard operating procedures (SOPs) were approved by the Institutional Animal Care and Use Committee (IACUC) and the Division of Laboratory Animal Resources (DLAR) at Duke University Medical Center (DUMC).
Test/control article preparation and quality control
A stock formulation of D2C7-IT was diluted in a control formulation, which consisted of phosphate buffer, saline, human serum albumin (HSA) (USP; Grifols, Clayton, NC, USA) or rat serum albumin (RSA) (catalog # RTSA62-1256; Equitech-Bio, Inc., Kerrville, TX, USA), 1 mM gadolinium [Gd-DTPA; Magnevist (gadopentetate dimeglumine); Bayer Health Care, Whippany NJ, USA], and 2 μCi 124I-labeled (IBA Molecular, Dulles, VA, USA) HSA (Talecris Biotherapeutics, Research Triangle Park, NC, USA) [124I-HSA, labeled in house and evaluated by size exclusion high performance liquid chromatography (HPLC)], on each day of use to yield formulations containing D2C7-IT at different concentrations. Before the initiation of the study, biochemical analysis was performed to ensure the strength, purity, and identity of the active ingredients in the test article formulation. Aliquots were immediately collected for each group after formulation and tested for the potency measurement, sterility, and endotoxin level. As for the potency measurement, formulated D2C7-IT samples for each set were either transferred to 15-ml sterile tubes (the bulk test article) or used to fill the osmotic pumps (the pump test article). The filled osmotic pumps were then placed into sterile 50-ml tubes containing sterile saline, which covered the pumps. The bulk test article and pump test article were incubated at 37 °C for the subsequent measurements of the inhibition (the half maximal inhibitory concentration, IC50) of A431P (EGFRwt expressing) and/or NR6M (EGFRvIII expressing) cell proliferation [11]. A cytotoxicity assay was performed following the CellTiter-Fluor Cell Viability Assay protocol (Promega, Madison, WI, USA). The cytotoxic activities of the pump samples were compared to those of the bulk samples that were used as reference standard controls in this assay. In addition, trypticase soy broth (BD, Sparks, MD, USA) and fluid thioglycollate medium (BD, Sparks, MD, USA) were utilized to test the sterility of each formulation sample, whereas the limulus amebocyte lysate (LAL) test (Endosafe®; Charles River, Charleston, SC, USA) was used to determine the endotoxin level of the formulation samples.
Intracerebral CED by osmotic pump
An osmotic pump (Alzet Osmotic Pumps 2ML1 or 2001; Durect Corporation, Cupertino, CA) was connected to a brain infusion cannula (Alzet Brain Infusion Kit 2; Durect Corporation, Cupertino, CA) to form a pump assembly. The control or test article formulations were prepared fresh each day, loaded into the pump assembly, maintained at 37 °C overnight to facilitate pump priming, and implanted into each rat for intracerebral infusion over approximately 72 h. The implantation procedure for the osmotic pump assembly was as follows: anesthesia was induced and maintained with isoflurane (Henry Schein Animal Health, Dublin, OH, USA); the hair over the surgical site was clipped, cleaned with alcohol (70 % isopropyl alcohol; Priority Care, Inc., Elgin, IL, USA) and scrubbed with Avagard (1 % chlorhexidine gluconate, Avagard Surgical and Health Care; 3 M, St. Paul, MN, USA); while anesthetized, the animals were placed into a stereotaxic frame; the intracerebral cannula was placed 1 mm anterior to the bregma, 3 mm to the right of the cranium midline, and 5 mm into the caudate nucleus (Fig. 1a and b), which was held in place using cyanoacrylate adhesive (3 M Vetbond Tissue Adhesive; 3 M Animal Care Products, St. Paul, MN, USA); the primed pump was implanted subcutaneously into the mid-scapular region of each animal; and the incision was closed with sterile wound clips (MikRon Precision Inc., Gardena, GA, USA). The beginning of the infusion period was designated as the time when the cannula/pump assembly was placed into the brain. The infusion pump was removed from each animal approximately 72 h after the start of the infusion, and the cannula was left in the brain.

a The coordinates of the cannula in the rat brain (the coronary view). b The diagram of the CED pump system used in the intracerebral infusion (the sagittal view)
One-month toxicity study of SD rats
Two cohorts of SD rats were evaluated for systemic toxicity (acute cohort: necropsy on study day 5, and chronic cohort: necropsy on study day 34). Each cohort consisted of four groups (control, low, medium, and high dose of D2C7-IT) consisting of five male rats and five female rats per dose group. All animals were observed upon receipt into the facility, and daily observations of each animal were performed and recorded throughout the study for moribundity, mortality, and general well-being. In addition, the animals were thoroughly examined for pharmacotoxic signs, pain, or gross abnormalities on the day before pump implantation and then once weekly throughout the study until the surviving animals were humanely euthanized. The rats in both cohorts (acute and chronic) were weighed on study day 1 (before implantation), study day 5, weekly thereafter (for surviving animals in the chronic cohort), and before euthanasia. Blood was collected from each rat for the clinical laboratory tests, including hematology, serum chemistry, and blood cell morphology (ANTECH Diagnostics, Morrisville, NC, USA) before euthanasia on days 5 or 34, or before unscheduled euthanization of moribund rats. On the scheduled study termination dates and following the unscheduled euthanization of moribund rats, the animals were euthanized for complete necropsy. The animals found dead were necropsied upon discovery. At necropsy, major organ systems and tissues were collected, weighed, and then examined for gross lesions as well as for histopathologic changes.
Functional observation battery (FOB)
The FOB parameters were measured within one week before randomization into the study groups and for the surviving animals on study days 4, 15, and 31 (days 15 and 31 for the rats scheduled for necropsy on day 34 only) at the Mouse Behavioral and Neuroendocrine Analysis Core Facility at the Duke University Medical Center. The FOB was designed and validated for sensitive determination of neurobehavioral toxicity [15]. The FOB components included observations of home cage behaviors [convulsions (tonic and clonic) and vocalizations], and reactivity of the animals to handling by the investigator [ratings of the ease of removal of animals from cages (including aggressiveness), coordination of the rat during handling, lacrimation, piloerection, and salivation]. Open-field activity measurements included the distance moved during testing and the time spent in mobility. Assessment of gait was performed in Clever Sys Free Walk automated test arenas with behavioral recognition software (Clever Sys Inc., Reston, VA, USA). Stimulus reactivity measurements to an auditory “click” and the core body temperature were also assessed. Behavioral data were analyzed by repeated measures ANOVA (RMANOVA) and chi-square analysis (χ 2) using IBM SPSS 21 statistical software (IBM, Chicago, IL, USA). In all cases, p < 0.05 was considered to be statistically significant.
Results
TOX-2013-001 preclinical toxicity trial
As shown in Table 1, the toxicity of D2C7-IT was investigated in four dose groups (0, 0.30, 0.50, and 0.63 μg/rat) in the acute and chronic cohorts. The initial control formulation, which was recommended by the FDA to regulate the human clinical formulation, consisted of 0.05 M PBS in saline, 0.2 % HSA, 2 μCi 124I-HSA, and 1 mM Gd-DTPA. The dose formulations were delivered into the right caudate nucleus of each individual rat via a subcutaneously implanted osmotic pump (Alzet pump 2ML1) [13] at a nominal 10.1 μL/h flow rate. Five rats per sex per treatment group were scheduled for necropsy on days 5 (the acute cohort) and 34 (the chronic cohort) of the study to examine the acute and chronic toxicities of the infused test article.
General adverse clinical signs, such as head-tilt, trembling, and loss of coordination, were observed among some animals in all of the groups of both cohorts (8/20 rats in the control group, 10/20 rats in the 0.3 μg dose group, 12/20 rats in the 0.5 μg dose group, and 14/20 rats in the 0.63 μg dose group). Unscheduled mortality was observed in 2/20 rats in the 0.3 μg dose group and 3/20 rats in the 0.63 μg dose group. Behavioral abnormalities, including the inability to walk or stand unassisted, abnormal posture, seizures, catalepsy, abnormal body temperature, and abnormal nasal or eye discharge were observed in all groups. At necropsy, no treatment-related gross lesions were observed. Histopathologic evaluation of the brains revealed regions of encephalomalacia (ECM) largely centered in the centrum semiovale area, which corresponded to the injection site, in all groups. The injection site was surrounded by haloes of gliosis and demyelination in all cases.
The pump D2C7-IT test formulations under the same conditions showed an approximately 10-fold loss of biological activity compared to the bulk samples (reference standard control), as measured by the inhibition of A431P (24–96 h IC50: Bulk 57–71 pg/mL vs Pump 750–850 pg/mL) and NR6M (24–96 h IC50: Bulk 490–600 pg/mL vs Pump 5900–7500 pg/mL) cell proliferation (Tables 2 and 3), suggesting formulation issues with the test article. Thus, the trial was terminated after approximately one week of the in-life portion. In addition, the dose formulations (osmolality of approximately 370 mOsm/l H2O) were proven to be hypertonic compared with the osmolality of the rat brain (290–300 mOsm/l H2O).
TOX-2013-002 preclinical toxicity trial
TOX-2013-002 was initiated with a modified isotonic dose formulation using sodium phosphate buffer in saline with 3 % HSA, 2 μCi 124I-HSA, 1 mM gadolinium, and D2C7-IT administered at four doses of 0, 0.075, 0.15, and 0.3 μg/rat over 72 h using a subcutaneously implanted osmotic pump (Alzet pump 2ML1) at a nominal 10.1 μL/h flow rate. General adverse clinical signs, including weight loss, dehydration, and seizure-like activity, were observed among many animals from both cohorts in the control group (13/20 rats) and the test article groups (17/20 rats in the 0.075 μg dose group, 14/20 in the 0.15 μg dose group, and 14/20 in the 0.3 μg dose group). Unscheduled mortality was observed in 4/20 rats in the control group, 4/20 in the 0.075 μg dose group, 3/20 in the 0.15 μg dose group, and 3/20 in the 0.3 μg dose group (Table 4). Behavioral assessments revealed abnormalities in all groups of both cohorts, including the inability to stand or walk unassisted, abnormal posture, continuous rotation, stereotypies, seizures, catalepsy, abnormal body temperature, and eye/nasal discharge. At necropsy, no treatment-related gross lesions were present. Evaluation of brain histopathology revealed regions of ECM (destructive lesions) largely centered in the centrum semiovale corresponding to the injection site in all study groups, including the control group (no dose–response relationships were noted among the injection site injuries). The areas of ECM were surrounded by haloes of gliosis and demyelination that were not dose-related in all groups. Demyelination, gliosis, and hydrocephalus were noted in all four groups. Although there was not a clear dose–response relationship, a large area (5.0 × 2.5 mm or greater) of ECM occurred more frequently in the groups administered with the test article.
Kidney histopathology revealed a similar number of lesions in the control and test article dose groups, including acute tubular necrosis, foci of lymphocytes and inflammation, and areas of calcification. None of these lesions occurred in a dose-related manner. Due to these abnormal clinical observations of the rats in all dose groups, including the control group, the study was terminated after 10 days of the in-life portion.
Vehicle toxicity trial
A new control formulation (sodium phosphate buffer in saline with 2 % RSA) was tested to evaluate its toxicity. The dose formulation was administered intracerebrally into the rat brain over 72 h using the smaller osmotic pump (Alzet pump 2001) at a slower nominal 1 μL/h flow rate. The dose formulation was delivered into the right (Group 1) or left (Group 2) caudate nucleus of individual rats in two groups consisting of five male rats and five female rats per group, and necropsy occurred on study day 7 for both groups. Clinical observations, body weight changes, and gross and microscopic pathologies were evaluated over a 7-day period in this trial. No apparent study-related adverse clinical signs were observed during the study. There were no obvious study-related differences in body weights between male or female rats in both groups. Microscopic histopathology showed normal brains in 13/20 rats, and the remaining 7 rats only displayed needle and catheter tracks and a slight degree of inflammation at the point of needle and catheter insertion. Because the rats in the vehicle toxicity trial showed better outcomes with this new control formulation, the HSA in the previous control formulation was replaced with RSA in the final D2C7-IT GLP toxicity trial.
TOX-2014-001 GLP preclinical toxicity trial
A final TOX-2014-001 GLP toxicity trial was performed to evaluate the acute and chronic toxicities of D2C7-IT administered via intracerebral CED to determine the maximum tolerated dose (MTD) and the no-observed-adverse-effect-level (NOAEL) in rats. D2C7-IT was formulated in an isotonic control formulation (potassium phosphate buffer in saline with 2 % RSA, 2 μCi 124I-HSA and 1 mM gadolinium). Dose formulations were delivered into the right caudate nucleus of individual rats via subcutaneously implanted osmotic pumps (Alzet pump 2001) at a nominal 1.01 μL/h flow rate. Each dose preparation passed the bacteria and endotoxin tests.
As summarized in Table 5, the test article was administered into five male rats and five female rats (total doses: 0, 0.05, 0.10, and 0.40 μg/rat) that underwent necropsy on study day 5 (the acute cohort) and five male rats and five female rats (total doses: 0, 0.05, 0.10, and 0.35 μg/rat) that underwent necropsy on study day 34 (the chronic cohort). Necropsy was completed in the acute cohort before the chronic cohort. Mortality occurred in 5/10 rats (3/5 males and 2/5 females) in the high-dose group (0.40 μg total dose) in the acute cohort. Consequently, the total dose was decreased from 0.40 to 0.35 μg for the high-dose group in the chronic cohort. Mortality was observed in 4/10 rats (2/5 males and 2/5 females) in the 0.35 μg total dose group in the chronic cohort.
There were no apparent study-related adverse clinical signs in either the acute and chronic cohort, except in the high-dose groups, in which weight loss, reluctance to ambulate, and difficulty in ambulating were observed in the rats that received either the 0.35 or 0.40 μg total dose. Seizure-like activities and moribund conditions were observed in the rats that received the 0.35 μg total dose in the chronic cohort. All study-related adverse clinical signs were noted on study days 4 to 6. There were no dose-related differences in body weight for male or female rats among the groups in the acute or the chronic cohort. Likewise, the effects of treatment over time on body weight did not differ among the groups of males or females in either cohort. There were no dose-related effects observed in terms of hematology, serum chemistry, or blood cell morphology in the males or females in either cohort.
The FOB conducted on the acute cohort on study day 4 for the 5-day study revealed that the male rats treated with the high dose of D2C7-IT displayed abnormal behaviors, including impaired exploration of the open field (Supplementary Figs. S1-2), which was secondary to gait and posture problems (Supplementary Figs. S3-4). Rats that were treated with 0.05 or 0.10 μg total doses of D2C7-IT, however, exhibited few changes. In addition to activity and gait, rats were assessed for general appearance, reactivity to handling, reaction to a single auditory “click,” and core body temperature on day 4 of the 5-day assessment. No statistically significant overall effects were found for general appearance and reactivity to handling of the rats. Following surgery, 33.3 % of rats in the 0.40 μg treatment group failed to exhibit a startle response (see supplemental data). For core body temperature, no effects of treatment were detected for females (Supplementary Fig. S5). By comparison, males given 0.40 μg D2C7-IT had a significant reduction in post-surgical relative to pre-surgical body temperatures (see supplemental data). Together, these data show that by day 4 of the 5-day study, male rats given the highest dose of D2C7-IT were more likely to have reductions in activity secondary to abnormalities in gait, reduced startle reactivity, and decreased core body temperatures. By comparison, female rats remained relatively unaffected with this treatment.
The rats in the D2C7-IT high-dose groups during both the 5- and 34-day studies showed similar behavioral changes during the FOB. However, in the 34-day study, many of the behavioral abnormalities observed on day 4 in the chronic cohort were not evident on study days 15 and 31, or they had declined in prevalence or severity. In the open field, female rats showed no marked effects of treatment on activity, whereas males administered 0.35 μg D2C7-IT failed to exhibit habituation to the open field across repeated exposures (Supplementary Fig. S6). Furthermore, males that received 0.10 or 0.35 μg compound spent more time in low mobility on days 15 and 31 relative to females undergoing the same treatments (Supplementary Fig. S7). An investigation of gait during open field testing found the treatment effects were similar to those observed in the 5-day study (Supplementary Figs. S8-9). The male rats treated with D2C7-IT exhibited changes in open field activity, which may be related to gait alterations. During the 34-day study, a few other observations were notable, including salivation/drooling, changes in the coloration of the oral tissues and gums, coordination of the animal during handling, and startle reactivity to an auditory “click” stimulus. In each case, the effects of D2C7-IT were mild and only temporary, being fully resolved over the 34 days (see supplemental data). Finally, no marked effects of D2C7-IT were found on core body temperature in the 34-day study. Taken together, the observations in the 34-day study support the findings of the short-term study. Behavioral abnormalities with D2C7-IT were observed in the animals treated with the highest doses of D2C7-IT and they were most pronounced in males. Despite these results, the behavioral effects of D2C7-IT were transient, and were not observed by the end of the 34-day study.
With regard to brain histopathology, the animals in the acute cohort exhibited features consistent with dose-related injuries, which were confined to those in the high-dose group. Edema (8/10 rats), ECM (9/10), and demyelination (2/10 rats) were more widely distributed and more frequently found in the high-dose group than in the other groups of the acute cohort (Table 5 and Fig. 2). Among the animals in the high-dose group of the chronic cohort, a less obvious dose-related response was noted, with ECM, edema, and demyelination present in 7/10, 2/10, and 4/10 rats, respectively (Table 5). ECM was present at the injection site in the control group (10/20), low-dose group (7/20), and medium-dose group (7/10) of both cohorts (Table 5). However, edema and demyelination were not observed in the control, low-, and medium-dose groups of either cohort (Table 5). Pathologic examination of the systemic tissues from the animals in both cohorts revealed no dose-related or drug-related findings.

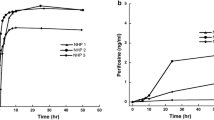
a A brain histopathologic picture shows that the encephalomalacia (ECM, within the red circle, 10X) occurred in the rats from the high dose group. b The D2C7-IT dose–response curve demonstrates that the LD40 dose of the D2C7-IT was 0.35 μg over 72 h, while the anticipated LD10 dose lay between 0.1 and 0.35 μg over a 72-h CED delivery
The MTD of D2C7-IT administered by intracerebral CED over a 72-h period was between 0.10 and 0.35 μg total dose (Fig. 2b). Considering the histopathologic and clinical findings, we conclude that the 0.05 μg total dose is the appropriate NOAEL for D2C7-IT in rats.
Discussion
Glioblastoma, the most malignant brain tumor, has the poorest prognosis, with a median survival time of less than 2 years. There is an urgent need for the development of advanced novel therapies to treat this fatal disease. D2C7-IT is a novel targeted IT developed in our laboratory. D2C7-IT has high affinity and specificity for both the EGFRwt and EGFRvIII proteins, it exhibits robust cytotoxic activities against glioblastoma cells expressing EGFRwt, EGFRvIII, or both (in vitro), and it significantly improves (>150 %) the survival of orthotopic mouse glioma models [11, 12]. Because the intravenous or oral delivery of macromolecules into the CNS is impeded by the blood–brain barrier (BBB), especially with large soluble molecules such as antibodies or ITs [16], intratumoral administration of D2C7-IT was performed in our animal model by a CED method, in which we used an osmotic pump to deliver the protein directly into the brain [12, 13]. Intracerebral CED for IT delivery has been well established and has shown promise [17–19]. Co-infusion of Gd-DTPA (a small-molecule contrast agent) and 124I-HSA has enabled the uses of MRI and PET scanning to accurately display imaging of the intracerebral distribution of an infused IT administered by CED. Co-infusion of Gd-DTPA and 124I-HSA has been demonstrated to be a safe and minimally invasive technique for patients [14].
A preclinical rat GLP study was performed to evaluate the systemic toxicity of intracerebral CED of D2C7-IT in a formulation containing Gd-DTPA and 124I-HSA, because co-infusion of these tracers will be used to monitor D2C7-IT intracerebral delivery and distribution in patients. Several preclinical toxicity trials were performed to determine the MTD and NOAEL as references for the initial dosing in our Phase I clinical trial. Nevertheless, numerous unexpected obstacles emerged during these trials, including the osmolality of the dose formulation, the potential absorption of the IT into the interior reservoir of the osmotic pump, non-optimal pump flow rate for the rat brain, and potential immunogenicity and adverse effects of HSA in the formulation.
Osmolality of the dose formulation
As summarized in Table 1, abnormal clinical signs were observed in animals in all dose groups in the TOX-2013-001 trial. Histopathologic lesions (including ECM, gliosis, and demyelination) were detected in the brains of animals in all dose groups, including the control group. Because all animal groups were equally affected, the control formulation, which was a common parameter across all groups, was assessed for potential flaws. Osmolality is a critical factor for agents delivered into the CNS [20, 21]. The osmolality of a normal rat’s CNS is 290–300 mOsm/l H2O. In the TOX-2013-001 trial, 0.05 M PBS-0.9 % saline was used in the control formulation to dilute D2C7-IT, Gd-DTPA, and 124I-HSA, because it is widely used clinically as a diluent. However, the osmolality of the 0.05 M PBS-0.9 % saline solution was approximately 370 mOsm/l H2O, which was more hypertonic than a normal rat’s CNS. The hypertonicity of the formulation could have been one of the major factors causing the adverse events observed in all groups. Therefore, in subsequent trials, the osmolality of the dose formulation was adjusted to 290–300 mOsm/l H2O with an appropriate concentration of phosphate buffer (Tables 4 and 5).
Potential adsorption of the IT into the interior reservoir of the osmotic pump
In the TOX-2-13-001 trial, the pumped-out D2C7-IT test formulation showed an approximately 10-fold loss of biological activity compared with the bulk D2C7-IT under the same conditions based on the inhibition of A431P and NR6M cell viability measured after 48 h of incubation (Tables 2 and 3, respectively). This finding suggested that the concentration of D2C7-IT in the pumped-out solution was much lower than that in the bulk solution. Because a 1–5 % concentration of the carrier protein was recommended by the pump manufacturer to be included in the formulation to minimize test article (D2C7-IT) adsorption in the osmotic pump, the HSA concentration was increased from 0.2 to 0.3 % to prevent D2C7-IT adsorption into the interior reservoir of the pump. The increased HSA concentration in the formulation restored the in vitro cytotoxic activity of the pumped-out D2C7-IT (in 3 % HSA solution) (data not shown). As a result, a 3 % HSA concentration was used in the dose formulation in the TOX-2013-002 trial.
Optimal pump flow rate for the rat brain
Adverse events, body weight loss, and unscheduled mortality were again observed after intracerebral delivery of the new control formulation in all treatment groups during the TOX-2013-002 trial. Brain histopathology revealed regions of ECM largely centered in the centrum semiovale that corresponded to the injection site in all study groups. The observed abnormalities could have developed because the volume of the pumped out formulation was too high for the rat brain. Based on the results of a previous IT preclinical toxicity study, an Alzet pump 2ML1 (size: 2 ml; duration: 7 days) was used in the TOX-2013-001 and TOX-2013-002 trials to continuously deliver the formulations into the rat brain at a flow rate of 10 μL/h for 72 h [13, 22]. However, upon consultation with the pump manufacturer and senior investigators from the Department of Psychiatry and Behavioral Sciences at Duke University, who have extensive experience with the use of osmotic pumps in preclinical animal models, a lower flow rate (e.g., less than 5 μL/h) was suggested to be preferential for administering compounds into the rat brain parenchyma. Therefore, in subsequent trials, we switched to a smaller Alzet pump 2001 (size: 200 μl; duration: 7 days) with a slower flow rate of 1 μL/h for intracerebral infusion.
Potential HSA immunogenicity and adverse effects in rats
A second unexpected finding of the TOX-2013-002 trial was the occurrence of kidney lesions after infusion. Kidney histopathology revealed a similar number of lesions in the control and test article dose groups, including acute tubular necrosis, foci of lymphocytes and inflammation, and areas of calcification. None of these lesions occurred in a dose-related manner, implying that a common component of the control and test articles was responsible for the abnormal findings in the kidney. Because its concentration was increased from 0.2 to 3 %, the total amount of HSA infused into the CNS was nearly 22 mg per rat over the 72-h period. Harling-Berg and colleagues [23] have previously reported efficient immunization of rats with only 90 μg HSA microinfused into the rat’s lateral ventricle. Moreover, HSA has been demonstrated to be more immunogenic when microinfused into cerebrospinal fluid (CSF) than when injected intravenously, intramuscularly, or intraperitoneally. HSA antigenicity leads to the formation of soluble circulating immune complexes, which can result in glomerular injury [24]. Possible HSA immunogenicity may explain the kidney lesions found in all treatment groups, including the control group. The HSA formulation was actually used to accurately replicate the intended human clinical formulation; however, after demonstrating the potential toxicity of HSA in rats, the FDA agreed to substitute RSA for HSA.
A satisfactory outcome was observed with the uses of the modified control formulation containing 2 % RSA and a smaller osmotic pump with a slower flow rate, which was used in the vehicle-only trial. Unfortunately, right after the vehicle test trial, the sodium phosphate injection (Hospira, Lake Forest, IL) used in the dose formulation was nationally back ordered. Upon consultation with the Duke Medical Center pharmacist, potassium phosphate injection was utilized as an alternative to sodium phosphate buffer for the official GLP trial (TOX-2014-001). After successful completion of the TOX-2014-001 trial, the MTD of the D2C7-IT administered via intracerebral CED over a 72-h period was determined to be between a total dose of 0.1 and 0.35 μg and the NOAEL was a total dose of 0.05 μg in SD rats. Based on this successful GLP toxicity study, an IND is now in effect for a Phase I/II D2C7-IT clinical trial (NCT02303678, D2C7 for Adult Patients with Recurrent Malignant Glioma, clinicaltrials.gov), and patients with recurrent glioblastomas have been treated without obvious toxicity at the doses of 40, 80, and 120 ng of D2C7-IT administered by CED intratumorally for 72 h.
Conclusions
In this preclinical GLP study, the systemic toxicity of a novel IT, D2C7-(scdsFv)-PE38KDEL, was examined in a rat intracerebral CED model over a 72-h period. Several critical issues emerged during the first two trials, including the osmolality of the dose formulation, the pump flow rate, the adsorption of the IT into the pump interior reservoir, and the HSA immunogenicity and adverse effects in rats. These issues were addressed by correcting the osmolality of the dose formulation, selecting a smaller osmotic pump with a slower flow rate, and substituting 2 % RSA for 3 % HSA. Ultimately, the MTD of D2C7-IT was determined to be between a total dose of 0.10 and 0.35 μg, and the NOAEL of D2C7-IT was a total dose of 0.05 μg in SD rats. Both the MTD and NOAEL were utilized as references for the D2C7-IT clinical trial design.
References
Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J (2014) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol 16(Suppl 4):iv1–iv63. doi:10.1093/neuonc/nou223
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359(5):492–507. doi:10.1056/NEJMra0708126
Chandramohan V, Sampson JH, Pastan I, Bigner DD (2012) Toxin-based targeted therapy for malignant brain tumors. Clin Dev Immunol 2012:480429. doi:10.1155/2012/480429
Ahmad ZA, Yeap SK, Ali AM, Ho WY, Alitheen NB, Hamid M (2012) scFv antibody: principles and clinical application. Clin Dev Immunol 2012:980250. doi:10.1155/2012/980250
Shapira A, Benhar I (2010) Toxin-based therapeutic approaches. Toxins 2(11):2519–2583. doi:10.3390/toxins2112519
Pastan I, Hassan R, Fitzgerald DJ, Kreitman RJ (2006) Immunotoxin therapy of cancer. Nat Rev Cancer 6(7):559–565. doi:10.1038/nrc1891
Piao H, Kuan CT, Chandramohan V, Keir ST, Pegram CN, Bao X, Mansson JE, Pastan IH, Bigner DD (2013) Affinity-matured recombinant immunotoxin targeting gangliosides 3′-isoLM1 and 3′,6′-isoLD1 on malignant gliomas. MAbs 5(5):748–762. doi:10.4161/mabs.25860
Salomon DS, Brandt R, Ciardiello F, Normanno N (1995) Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 19(3):183–232
Chaffanet M, Chauvin C, Laine M, Berger F, Chedin M, Rost N, Nissou MF, Benabid AL (1992) EGF receptor amplification and expression in human brain tumours. Eur J Cancer 28(1):11–17
Frederick L, Wang XY, Eley G, James CD (2000) Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res 60(5):1383–1387
Chandramohan V, Bao X, Keir ST, Pegram CN, Szafranski SE, Piao H, Wikstrand CJ, McLendon RE, Kuan CT, Pastan IH, Bigner DD (2013) Construction of an immunotoxin, D2C7-(scdsFv)-PE38KDEL, targeting EGFRwt and EGFRvIII for brain tumor therapy. Clin Cancer Res 19(17):4717–4727. doi:10.1158/1078-0432.CCR-12-3891
Bao X, Chandramohan V, Keir ST, Pegram CN, McLendon RE, Kuan C-T, Pastan IH, Bigner DD (2013) Antitumor efficacy of D2C7-(scdsFv)-PE38KDEL, a novel immunotoxin targeting EGFRwt and EGFRvIII, by convection-enhanced delivery in orthotopic brain tumor mouse models. J Immunother Cancer 1(Suppl 1):P126. doi:10.1186/2051-1426-1-s1-p126
Ding D, Kanaly CW, Bigner DD, Cummings TJ, Herndon JE 2nd, Pastan I, Raghavan R, Sampson JH (2010) Convection-enhanced delivery of free gadolinium with the recombinant immunotoxin MR1-1. J Neuro-Oncol 98(1):1–7. doi:10.1007/s11060-009-0046-7
Sampson JH, Brady M, Raghavan R, Mehta AI, Friedman AH, Reardon DA, Petry NA, Barboriak DP, Wong TZ, Zalutsky MR, Lally-Goss D, Bigner DD (2011) Colocalization of gadolinium-diethylene triamine pentaacetic acid with high-molecular-weight molecules after intracerebral convection-enhanced delivery in humans. Neurosurgery 69(3):668–676. doi:10.1227/NEU.0b013e3182181ba8
Moser VC (1991) Applications of a neurobehavioral screening battery. Int J Toxicol 10(6):661–669. doi:10.3109/10915819109078658
Blanchette M, Fortin D (2011) Blood–brain barrier disruption in the treatment of brain tumors. Methods Mol Biol 686:447–463. doi:10.1007/978-1-60761-938-3_23
Sampson JH, Akabani G, Archer GE, Berger MS, Coleman RE, Friedman AH, Friedman HS, Greer K, Herndon JE 2nd, Kunwar S, McLendon RE, Paolino A, Petry NA, Provenzale JM, Reardon DA, Wong TZ, Zalutsky MR, Pastan I, Bigner DD (2008) Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro Oncol 10(3):320–329. doi:10.1215/15228517-2008-012
Mehta AI, Choi BD, Ajay D, Raghavan R, Brady M, Friedman AH, Pastan I, Bigner DD, Sampson JH (2012) Convection enhanced delivery of macromolecules for brain tumors. Curr Drug Discov Technol 9(4):305–310
Sampson JH, Raghavan R, Brady M, Friedman AH, Bigner D (2011) Convection-enhanced delivery. J Neurosurg 115(3):463–464. doi:10.3171/2010.11.JNS101801, discussion 465–466
Kawamata T, Mori T, Sato S, Katayama Y (2007) Tissue hyperosmolality and brain edema in cerebral contusion. Neurosurg Focus 22(5):E5
Bandaranayake NM, Nemoto EM, Stezoski SW (1978) Rat brain osmolality during barbiturate anesthesia and global brain ischemia. Stroke 9(3):249–254. doi:10.1161/01.str.9.3.249
Noker PE, Fulton R, Pickett AC, Mann JF (2007) Multiple dose toxicity study of MR1-1-(MR1-1dsFvPE38KDEL, NSC-718877) in Sprague Dawley rats. Conducted at Southern Research Institute, Birmingham, Alabama and sponsored by National Cancer Institute, Bethesda, Maryland
Harling-Berg C, Knopf PM, Merriam J, Cserr HF (1989) Role of cervical lymph nodes in the systemic humoral immune response to human serum albumin microinfused into rat cerebrospinal fluid. J Neuroimmunol 25(2–3):185–193
Palestro G, Mazzucco G, Navone R, Canese MG, Coda R, Novero D, Micca FB, Leonardo E (1980) Role of the T-cell system in glomerulonephritis induced in rats by human serum albumin (HSA). An immunological and morphological study. Virchows Arch B Cell Pathol Incl Mol Pathol 35(1):19–32
Acknowledgments
We thank Terri Lucas and Lena Perdue for coordinating the study, Charles Pegram, David Soule, and Xiao-Guang Zhao for the formulation preparation, and Colleen Herbst, Meredith Weksler, and Fernando Orozco for their surgical assistance. We wish to thank Christopher Means and Theo Rhodes for their detailed assessment of the animals for the Functional Observation Battery (FOB) and assistance in preparing and analyzing the FOB data. We also thank Jenna Lewis for her editorial assistance. The study was funded by the following grant from the National Institutes of Health (NIH) of the United States: P01-CA154291-03 (to D.D. Bigner). This research was also supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Rights and permissions
About this article

Cite this article
Bao, X., Chandramohan, V., Reynolds, R.P. et al. Preclinical toxicity evaluation of a novel immunotoxin, D2C7-(scdsFv)-PE38KDEL, administered via intracerebral convection-enhanced delivery in rats. Invest New Drugs 34, 149–158 (2016). https://doi.org/10.1007/s10637-015-0318-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-015-0318-3