
Abstract
Purpose
Central nervous system tumors are histologically and biologically heterogeneous. Standard treatment for malignant tumors includes surgery, radiation and chemotherapy, yet surgical resection is not always an option and chemotherapeutic agents have limited benefit. Recent investigations have focused on molecularly targeted therapies aimed at critical tumorigenic pathways. Several tumor types, including high-grade gliomas and pediatric pontine gliomas, exhibit Akt activation. Perifosine, an orally bioavailable, synthetic alkylphospholipid and potent Akt inhibitor, has demonstrated activity in some preclinical models, but absent activity in a genetically engineered mouse model of pontine glioma. We evaluated the plasma and cerebrospinal fluid pharmacokinetics of orally administered perifosine in a non-human primate model to evaluate CNS penetration.
Methods
Perifosine was administered orally to three adult rhesus monkeys as a single dose of 7.0 mg/kg perifosine. Serial paired plasma and CSF samples were collected for up to 64 days. Perifosine was quantified with a validated HPLC/tandem mass spectrometry assay. Pharmacokinetic parameters were estimated using non-compartmental methods. CSF penetration was calculated from the areas under the concentration–time curves.
Results
Peak plasma concentrations (C max) ranged from 11.7–19.3 µM, and remained >1 µM for >28 days. Time to C max (T max) was 19 h. The median (range) AUCPl was 3148 (2502–4705) µM/h, with a median (range) terminal half-life (t 1/2) of 193 (170–221) h. Plasma clearance was 494 (329–637) mL/h/kg. Peak CSF concentrations were 4.1–10.1 nM (T max 64–235 h). CSF AUCs and t 1/2 were 6358 (2266–7568) nM/h and 277 (146–350) h, respectively. Perifosine concentrations in the CSF remained over nM for >35 days. The mean CSF penetration was 0.16 %.
Conclusion
CNS penetration of perifosine after systemic administration is poor. However, levels were measurable in both plasma and CSF for an extended time (>2 months) after a single oral dose.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Malignant gliomas in both adult and pediatric patients are associated with a poor prognosis. While some central nervous system (CNS) tumors, such as medulloblastomas, may have better prognoses, treatment is associated with significant neurotoxicity. New treatment strategies that are more efficacious and specific for tumor cells are needed. The availability of molecularly targeted agents has prompted investigations into tumor biology in efforts to better understand and target specific pathways associated with tumor pathogenesis. One such pathway, PI3K/Akt/mTOR, has been implicated in a number of cancers, including brain tumors. Activation of this pathway has been demonstrated in ~70 % of glioblastomas [1], the majority of pediatric gliomas [2, 3], and medulloblastomas [4]. Akt (protein kinase B) is a serine/threonine kinase involved in regulation of proliferation, differentiation, metabolism, and cell survival [5]. Its activity is indirectly inhibited by phosphatase and tensin homolog (PTEN) via PI3K inhibition; loss of PTEN function, which is also observed in a number of brain tumors, can lead to Akt activation [6]. Although several inhibitors of this pathway, such as the mTOR inhibitor, everolimus [7, 8], are under clinical investigation for the treatment of glioma, specific upstream targeting of Akt is of interest to avoid potential upregulation of alternate pathways.
Alkylphospholipids, a class of antitumor agents which include the agent, perifosine, interact with the cell membrane rather than nuclear DNA, and block signal transduction pathways [9]. Perifosine (octadecylphosphopiperidine) is an oral synthetic alkylphosphocholine-like molecule that has demonstrated cytotoxic effects against a number of human tumor cell lines and selectivity against neoplastic versus normal hematologic cells in vitro [10, 11]. While many kinase inhibitors target the ATP-binding region of their respective kinase, perifosine inhibits Akt activation by targeting the pleckstrin homology domain of Akt, thus preventing translocation to the plasma membrane and subsequent phosphorylation [12]. Preclinical studies of perifosine have shown activity against a wide variety of human tumor cell lines, with reported IC.50 in the range of .09–9.2 µg/ml [13, 14]. Its effects appear to be dose- and schedule-dependent [15]. Unlike other agents in this class, perifosine has been relatively well tolerated in early phase clinical trials [16, 17].
Perifosine has been investigated in Phase I and Phase II clinical trials where single-agent objective responses have been reported in patients with renal cell carcinoma, sarcoma and Waldenstroms macroglobulinemia [12]. Plasma pharmacokinetics of perifosine in adult patients with solid tumors has been described [18]. Following an oral dose, plasma concentrations peaked at 8–24 h followed by prolonged disappearance of perifosine from the plasma with half-lives of 80–116 h.
There is interest in investigating perifosine for treatment of malignant gliomas given that the Akt pathway is frequently implicated in the pathogenesis of malignant gliomas including diffuse intrinsic pontine glioma (DIPG), perifosine has demonstrated activity in vitro against glioma cell lines [1] and in vivo in mice bearing U251 xenografts injected into the hind limb [19], and the agent is relatively well tolerated. However, in a mouse model of DIPG in which the PI3Kinase/Akt pathway is activated, no activity was observed in response to perifosine treatment [20]. We hypothesize that this is due to inadequate delivery to the CNS. Penetration into the cerebrospinal fluid (CSF) has not been evaluated. In this study, we investigated the plasma and CSF pharmacokinetics of perifosine after a single oral dose in a non-human primate model previously shown to be predictive of CSF pharmacokinetics of many anticancer drugs in humans [21].
Materials and methods
Drug preparation
Perifosine was provided by the Cancer Therapy Evaluation Program (supplied to the National Cancer Institute under CRADA #0774 with Keryx Biopharmaceuticals, Inc.) in pure bulk compound. The drug was dissolved in normal saline and immediately administered as described below.
Animals
The National Cancer Institute Institutional Animal Care and Use Committee approved this study. Three adult male rhesus monkeys (Macaca mulatta) ages 13–15 years, weighing 10.5–14.7 kg, negative for SRV/SIV, and Herpes B viruses, were utilized. An indwelling, subcutaneous fourth ventricular catheter and reservoir system for CSF collection and a central venous port for blood collection were implanted in each animal. The animals were socially housed in groups, fed LabDiet Monkey Chow #5038 with water ad libitum, and cared for in accordance with the National Research Council (NRC) Guide for the Care and Use of Laboratory Animals [22].
Experiments
Animals were sedated (Ketamine, IM, 10 mg/kg, Vedco Inc., Saint Joseph, MO), and perifosine at a dose of 7 mg/kg (human equivalent dose 3.78 mg/kg) was administered orally via a nasogastric tube. Initially, blood (3 ml) and CSF (0.3 ml) were obtained at multiple time points between 0–10, 24, and 48 h. Subsequent studies were performed on the same animals, with blood and CSF collection extended beyond 48 h given the absence of detectable perifosine at early time points and the known prolonged terminal half-life of this agent; blood (3 ml) and CSF (0.3 ml) were obtained at 0, 1, 3, 6, 8, 10, 13, 14, 22, 28, 35, 42, 50 and 64 days following drug administration. Plasma was immediately separated by centrifugation. Plasma and CSF were stored at −20 °C then transferred to storage at −80 °C until analysis. Total drug concentrations of perifosine were quantified using a modification of a validated HPLC/tandem mass-spectrometry assay [23]. Briefly, plasma samples were spiked with a known amount of internal standard (miltefosine, Sigma-Aldrich, St. Louis, MO) and prepared by protein precipitation in acetonitrile (Mallinckrodt Baker, Phillipsburg, NJ). Experiments were carried out with a Shimadzu (Shimadzu Corp, Tokyo, Japan) HPLC system consisting of a controller module (CBM-20A), mobile phase vacuum degassing unit (DGU-20A3), binary pumps (LC-20AD), temperature-controlled autosampler (SIL 20-AC) and column oven (CTO-20AC). Analyte separation was achieved using a Devosil C30-UG (10 × 4 mm, 5 µm particle size) column (Nomura Chemical Company by Phenomenex, Torrance, CA) preceded with a KrudKatcher pre-column filter (Phenomenex) at 25 °C with isocratic elution at 0.5 ml/min using a mobile phase consisting of 9 mM ammonium formate (Sigma-Aldrich, Co., St. Louis, MO), pH 8, in 60 % acetonitrile. Miltefosine and perifosine eluted at 1.0 and 2.2 min, respectively. Compound detection was achieved using an API 5000 tandem mass spectrometer (ABI SCIEX, Foster City, CA) in positive ion turbo spray mode by following transitions 462–112 amu (perifosine) and 408–125 amu (miltefosine). Mass spectrometric data were analyzed using Analyst version 1.4.2 software (ABI SCIEX). NHP plasma concentrations were measured against standards made in pooled NHP plasma. The NHP CSF standard assay was validated using pooled NHP CSF, and in individual assays, pre-treatment CSF was used for quality control samples. Plasma standard curves were linear over a range of 0.001–10 µg/ml. Lower limits of quantification for perifosine in plasma and CSF were 1 and 0.1 ng/ml, respectively.
Pharmacokinetic analysis
Plasma and CSF pharmacokinetic parameters were estimated using non-compartmental methods. The peak perifosine concentration (C max) and time to peak concentration (T max) were determined from the time–concentration values for individual non-human primates. The area under the concentration–time curve (AUC) was calculated using the linear trapezoidal method with extrapolation of the curve to infinity (AUC0−inf). The terminal half-life was calculated by dividing 0.693 by the terminal rate constant. Clearance (Cl) was calculated by dividing the perifosine dose by the AUC0−inf. CSF penetration was calculated from the areas under the concentration–time curves of CSF and plasma.
Results
In the initial experiments where blood and CSF were collected up to 48 h, perifosine C max in the CSF had not been reached by 48 h (Fig. 1). The experiment was repeated with longer sampling times collected through 64 days after perifosine administration as noted above. Model-independent plasma pharmacokinetic parameters are shown in Table 1a. Following an oral dose of 7 mg/kg, the maximum mean measured plasma concentration (C max) of perifosine was 7.1 µg/ml (range 5.3–8.8), which corresponds to 13.3 µM (range 5.3–19.1). Due to sampling times in these experiments, time to maximum concentration was approximately 19 h after the oral gavage dose. Plasma exposure to perifosine as measured by the AUC was 1569 (1137–2139) µg h/ml. The terminal elimination half-life was calculated to be 195 (range 170–221) h (>8 days). Clearance is 59.1 (36.3–77.6) ml/h.

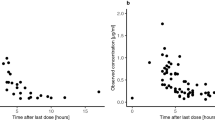
Plasma (a) and CSF (b) levels of perifosine in three nonhuman primates after oral dosing of 7.0 mg/kg and collection up to 48 h. C max and T max were reached by 48 h for plasma, but C max in CSF has not been reached by 48 h
CSF pharmacokinetic parameters are shown in Table 1b. The maximum measured CSF concentration of perifosine was 3.6 (1.85–4.57) ng/ml, which corresponds to 7.8 nM (range 4.0–9.9), and time to this maximum concentration was approximately 178 (63.6–235.4) h (>7 days), a long delay from the peak levels reached in plasma at approximately 19 h. CSF exposure to perifosine as measured by the area under the concentration–time curve is 2454 (1031–3440) ng h/ml. The terminal elimination half-life was calculated to be 258 (146–350) h (>10 days). The ratio of CSF to plasma exposure is .0016 (or .16 %).
The plasma and CSF concentration x time curves are shown in Fig. 2a, b, respectively. These curves demonstrate that absorption following oral dosing is slow. Disappearance of perifosine from plasma is multiphasic with a long terminal half-life. The appearance of perifosine in the CSF is delayed, with peak values reached 7 days after drug administration. While measurable drug is present in the CSF for up to 2 months, concentrations of perifosine in the CSF are 1000-fold less than plasma concentrations.

Plasma (a) and CSF (b) concentration × time curves of perifosine in non-human primates after extended sample collection up to 1500 h
Discussion
Perifosine is an Akt inhibitor with demonstrated activity in vitro but lacked activity in a genetically engineered mouse model of pontine glioma. A major reason for lack of efficacy of chemotherapeutic agents with demonstrated antitumor activity against CNS tumors in vitro is inadequate delivery to the tumor site, most likely due to the restrictive blood–brain barrier. This study demonstrates the limited penetration of the alkylphospholipid, perifosine, into the CSF relative to plasma drug exposure after gavage administration in nonhuman primates, with a ratio of CSF to plasma exposure of .0016 (or .16 %). As anticipated given the predictive pharmacokinetics in our non-human primate model, the plasma pharmacokinetic values are similar to those reported in adult patients with solid tumors, in whom the terminal half-life in humans was 105 h after dosing of 50–350 mg/day administered orally for 3 weeks [16].
Limited delivery to the CNS and, hence, limited delivery of antitumor agents to their site of action is a recurrent problem for the treatment of CNS tumors. All systemically administered chemotherapeutic agents must cross the capillary endothelial cells of the cerebral vasculature to gain access to tumor cells in the brain. Many hydrophilic substances and large lipophilic substances, including many chemotherapeutic compounds, are restricted from entering the CNS. The molecular weight of perifosine (462.66) likely restricts entry into the CNS. The limited penetration of perifosine could explain the lack of efficacy observed in a genetically engineered mouse model of pontine glioma despite observed activity in extracranial tumor models.
An intriguing pharmacokinetic characteristic demonstrated in this study is prolonged time to maximum concentration in the CSF after a single oral dose, and prolonged drug exposure due to a long terminal half-life (approximately 10 days) despite the poor CSF penetration of this agent when administered systemically. We were able to detect perifosine, albeit at low levels, in the CSF at 2 months following administration. Exposure to perifosine is extensive and suggests that the prolonged terminal half-life may make perifosine more suitable for local CNS administration, such as convection-enhanced delivery. Because of poor penetration across the blood–brain barrier, once perifosine is in the CNS, it should remain for long periods of time with little systemic exposure. It is possible that lower doses of perifosine could be administered directly to the CNS and result in higher perifosine exposure, increased Akt inhibition, and lower systemic toxicities compared to systemic administration.
In our studies, we use CSF as a surrogate of CNS tissue penetration. When using CSF penetration as a surrogate for CNS tissue penetration, several assumptions are made, including that there is a relationship between CSF levels and CNS tissue levels. For most agents, the correlation of drug exposure in the CSF versus that in the CNS tissue is not known, but previous studies of agents demonstrated comparable levels in brain extracellular fluid and cerebrospinal fluid when studied using microdialysis [24, 25].
In summary, this pharmacokinetic study of perifosine demonstrates poor CSF penetration of the agent after systemic administration but prolonged exposure due to extensive terminal half-life. Administration of perifosine directly into the CNS may be more efficacious for CNS tumors.
References
Momoto H, Nerio E, Holland E (2005) Perifosine inhibits multiple signaling pathways in glial progenitors and cooperates with temozolomide to arrest cell proliferation in gliomas in vivo. Cancer Res 65(16):7429–7435
Pollack I, Hamilton R, Burger P, Brat D, Rosenblum M, Murdoch G et al (2010) Akt activation is a common event in pediatric malignant gliomas and a potential adverse prognostic marker: a report from the children’s oncology group. J Neurooncol 99:155–163
Mueller S, Phillips J, Onar-Thomas A, Romero E, Zheng S, Wiencke J et al (2012) PTEN promoter methylation and activation of the PI3 K/Akt/mTOR pathway in pediatric gliomas and influence on clinical outcome. Neuro oncology 14(9):1146–1152
Kumar A, Fillmore H, Kadian R, Broaddus W, Tye G, Van Meter T (2009) The alkylphospholipid perifosine induces apoptosis and p21-mediated cell cycle arrest in medulloblastoma. Mol Cancer Res 7(11):1813–1821
Tokunaga E, Oki E, Egashira A, Sadanaga N, Morita M, Kakeji Y et al (2008) Deregulation of the Akt pathway in human cancer. Curr Cancer Drug Targets 8:27–36
Stambolic V, Suzuki A, de la Pompa J, Brothers G, Mirtsos C, Sasaki T et al (1998) Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95:29–39
Sarkaria J, Galanis E, Wu W, Peller P, Giannini C, Brown P et al (2011) North Central cancer treatment group phase I trial N057 K of everolimus (RAD001) and temozolomide in combination with radiation therapy in patients with newly diagnosed glioblastoma multiforme. Int J Radiat Oncol Biol Phys 81(2):468–475
Kreisl T, Lassman A, Mischel P, Rosen N, Scher H, Teruya-Feldstein J et al (2009) A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM). J Neurooncol 92(1):99–105
Kondapaka S, Singh S, Dasmahapatra G, Sausville E, Roy K (2003) Perifosine, a novel alkylphospholipid with a 462 m.w., inhibits protein kinase B activation. Mol Cancer Ther 2:1093–1103
Konstantinov S, Topaska-Ancheva M, Benner A, Berger M (1998) Alkylphosphocholines: effects on human leukemia cell lines and normal bone marrow cells. Int J Cancer 77(5):778–786
Bagley R, Jurtzberg L, Rouleau C, Yao M, Teicher B (2011) Erufosine, an alkylphosphocholine, with differential toxicity to human cancer cells and bone marrow cells. Cancer Chemother Pharmacol 68(6):1537–1546
Gills J, Dennis P (2009) Perifosine: update on a novel Akt inhibitor. Curr Oncol Rep 11:102–110
Schmidt-Heber M, Dadrowski R, Weimann A, Aicher B, Lohneis P, Busse A et al (2012) In vitro cytotoxicity of the novel antimyeloma agents perifosine, bortezomib and lenalidomide against different cell lines. Investig New Drugs 30(2):480–489
Li Z, Tan F, Liewehr D, Steinberg S, Thiele C (2010) In vitro and in vivo inhibition of neuroblastoma tumor cell growth by Akt inhibitor perifosine. J Natl Cancer Inst 102(11):759–770
Hennessy B, Lu Y, Poradosu E, Yu Q, Yu S, Hall H et al (2007) Pharmacodynamic markers of perifosine activity. Clin Cancer Res 13(24):7421–7431
Crul M, Rosing H, de Klerk G et al (2002) Phase I and pharmacological study of daily oral administration of perifosine (D-21266) in patients with advanced solid tumours. Eur J Cancer 38:1615–1621
Van Ummersen I, Binger K, Volkman J et al (2004) A phase I trial of perifosine (NSC 639966) on a loading dose/maintenance dose schedule in patients with advanced cancer. Clin Cancer Res 10:7450–7456
Unger C, Berdel W, Hanauske A, Sindermann H, Engel J, Mross K (2010) First-time-in-man and pharmacokinetic study of weekly oral perifosine in patients with solid tumours. Eur J Cancer 46(5):920–925
de la Pena L, Burgan W, Carter D, Hollingshead M, Satyamitra M, Camphausen K et al (2006) Inhibition of Akt by the alkylphospholipid perifosine does not enhance the radiosensitivity of human glioma cells. Mol Cancer Ther 5(6):1504–1510
Becher O, Hambardzumyan D, Walker R, Helmy K, Nazarian J, Albrecht S et al (2010) Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res 70(6):2548–2557
McCully C, Balis F, Bacher J, Phillips J, Poplack D (1990) A rhesus monkey model for continuous infusion of drugs into cerebrospinal fluid. Lab Anim Sci 40(5):520–525
Institute for Laboratory Animal Research (2011) Guide for the care and use of laboratory animals, 8th edn. National Academy Press, Washington (DC)
Woo E, Messman R, Sausville E, Figg W (2001) Quantitative determination of perifosine, a novel alkylphosphocholine anticancer agent, in human plasma by reverse-phase liquid chromatography-electrospray mass spectrometry. J Chromatogr B Biomed Sci Appl 759(2):247–257
Fox E, Bungay P, Bacher J et al (2002) Zidovudine concentration in brain extracellular fluid measured by microdialysis: steady-state and transient results in rhesus monkey. JPET 301:1003–1011
Jacobs S, McCully C, Murphy R et al (2010) Extracellular fluid concentrations of cisplatin, carboplatin, and oxaliplatin in brain, muscle, and blood measured using microdialysis in nonhuman primates. Cancer Chemother Pharmacol 65:817–824
Acknowledgments
This work was presented in part at the 2012 International Society of Pediatric Neuro-Oncology Meeting in Toronto. This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. The views herein do not necessarily represent the official views of the National Cancer Institute, the National Institutes of Health, the U.S. Department of Health and Human Services, or any other agency of the U.S. Government.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cole, D.E., Lester-McCully, C.M., Widemann, B.C. et al. Plasma and cerebrospinal fluid pharmacokinetics of the Akt inhibitor, perifosine, in a non-human primate model. Cancer Chemother Pharmacol 75, 923–928 (2015). https://doi.org/10.1007/s00280-015-2711-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-015-2711-1