
Summary
Low-dose metronomic chemotherapy is an emerging form of chemotherapy with distinct mechanisms of action from conventional chemotherapy (e.g., antiangiogenesis). Although developed to overcome resistance to conventional chemotherapy, metronomic chemotherapy is subject to resistance on its own. However, there is a paucity of information on mechanisms of resistance, on cross-resistance between metronomic regimens using different cytotoxic drugs, and on cross-resistance between metronomic versus conventional chemotherapy, or versus targeted antiangiogenic therapy. Herein we show that PC-3 human prostate cancer xenografts were sensitive to both metronomic cyclophosphamide and metronomic docetaxel, but resistant to metronomic topotecan. Conventional docetaxel was only moderately active in parental PC-3 and in metronomic cyclophosphamide resistant PC-3 tumors. However, in metronomic cyclophosphamide resistant PC-3 tumors combining conventional docetaxel or bolus cyclophosphamide therapy with continued metronomic cyclophosphamide was superior to each treatment alone. Furthermore, bevacizumab had single-agent activity against metronomic cyclophosphamide resistant PC-3 tumors. Microarray analyses identified altered regulation of protein translation as a potential mechanism of resistance to metronomic cyclophosphamide. Our results suggest that sensitivity to metronomic chemotherapy regimens using different cytotoxic drugs not only depends on shared mechanisms of action such as antiangiogenesis, but also on as yet unknown additional antitumor effects that appear to be drug-specific. As clinically observed with targeted antiangiogenic agents, the continued use of metronomic chemotherapy beyond progression may amplify the effects of added second-line therapies or vice versa. However, metronomic chemotherapy is no different from other systemic therapies in that predictive biomarkers will be essential to fully exploit this novel use of conventional chemotherapeutics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In order to maximize direct tumor cell kill, conventional chemotherapy is administered intermittently at the maximum tolerated dose, followed by treatment-free intervals to enable recovery of host tissues (e.g., bone marrow and intestinal mucosa) from therapy-induced toxic side-effects. In contrast, a different use of conventional chemotherapeutic agents has been described recently, i.e., low-dose metronomic chemotherapy, the frequent and uninterrupted long-term administration of small doses of cytotoxic drugs [1, 2]. Metronomic chemotherapy is characterized by the near-absence of acute high-grade side-effects, combined with promising antitumor effects seen in a number of phase II trials involving a broad range of tumor types, including studies of metronomic oral cyclophosphamide in prostate cancer [2–4]. Furthermore, the use of metronomic-like chemotherapy regimens improved overall survival in phase III trials of early lung and breast cancer [5, 6]. Since the majority of metronomic regimens are based on oral chemotherapeutics, metronomic chemotherapy is particularly suitable for outpatient use. Moreover, in view of the very beneficial toxicity profile, metronomic chemotherapy can be considered for elderly and frail patients that otherwise would not be candidates for conventional, maximum tolerated dose chemotherapy [7–10]. All in all, metronomic chemotherapy is emerging as a novel treatment alternative and is currently being clinically studied in at least six randomized phase III trials in either the adjuvant setting, in patients with advanced cancer, the elderly and frail, and as a maintenance treatment strategy following conventional induction chemotherapy [2, 11] (http://clinicaltrials.gov).
While the effects of conventional chemotherapy are primarily directed towards the cancer cell population, the antitumor activities of metronomic chemotherapy are thought to be mainly mediated through antiangiogenic effects [1]. In addition, recent evidence highlights the immunomodulatory potential of metronomic chemotherapy, whereas the importance of direct anti-cancer cell effects (e.g., interference with the hypoxia-induced factor 1α pathway, and targeting of cancer stem cells) remains to be determined [2, 4, 12, 13].
Initial preclinical studies by Browder et al. suggested that metronomic cyclophosphamide therapy is able to overcome resistance to conventional cyclophosphamide chemotherapy [14]. Furthermore, the antiangiogenic basis of metronomic chemotherapy would indicate that such regimens might be beneficial for the treatment of a broad range of tumor types. On the other hand, there is preclinical and clinical evidence showing intrinsic or acquired resistance to metronomic therapy [2, 3, 15, 16]. However, taking into consideration the different mechanisms of action of metronomic versus conventional chemotherapy, we speculated that likewise different mechanisms of resistance to metronomic versus conventional chemotherapy would be operative. In this regard, we recently showed in human prostate and breast cancer models that acquired resistance to metronomic cyclophosphamide chemotherapy is not associated with cross-resistance to maximum tolerated dose cyclophosphamide [15].
Herein we expanded on our previous studies by addressing three questions. First, does response to metronomic cyclophosphamide therapy predict response to metronomic regimens utilizing a different chemotherapeutic agent? Second, does in vivo acquired resistance to metronomic cyclophosphamide convey cross-resistance to second-line therapies, including to targeted antiangiogenic drugs such as vascular endothelial growth factor pathway inhibitors? Third, are there specific gene expression changes associated with resistance to metronomic cyclophosphamide?
Briefly, our results show that sensitivity to a metronomic regimen using a given drug (e.g., cyclophosphamide) does not necessarily predict response to another metronomic chemotherapy regimen utilizing a different chemotherapy drug (i.e., topotecan). Both treatment-naïve and metronomic cyclophosphamide resistant PC-3 prostate cancer xenografts responded poorly to maximum tolerated dose docetaxel, but maximum tolerated docetaxel combined with continued metronomic cyclophosphamide was superior compared to each treatment used alone in metronomic cyclophosphamide resistant tumors. In addition, metronomic cyclophosphamide resistant PC-3 xenografts responded well to anti-vascular endothelial growth factor antibody therapy using bevacizumab. Finally, altered regulation of protein translation appears to contribute to resistance to metronomic chemotherapy.
Materials and methods
Materials
Cyclophosphamide and docetaxel were obtained from the institutional pharmacy. Bevacizumab was kindly provided by Genentech Inc. (South San Francisco, CA), and oral topotecan was supplied by GlaxoSmithKline Inc. (Collegeville, PA). All drugs were reconstituted as per manufacturer’s instructions.
Animal procedures and treatment regimens
PC-3 human prostate cancer cells (ATCC, Manassas, VA) were maintained in a humidified atmosphere of 5 % CO2 at 37 °C in DMEM supplemented with 5 % fetal bovine serum. Metronomic cyclophosphamide resistant PC-3 cells (i.e., LCR1.1) and according control cells (i.e., NS1.1) were derived as previously described [15]. For tumor generation, 2 × 106 cells were injected subcutaneously into the flanks of 6–8 week old, 25–30 g male athymic nude mice (Harlan, Indianapolis, IN). Tumor size was assessed weekly using calipers and by applying the formula (0.5×[L × W2]), where L and W represent the largest and the smallest tumor diameter, respectively [17]. We obtained serial mouse body weights as a measure of treatment toxicity. All animal procedures were performed according to institutionally approved animal care guidelines (Sunnybrook Research Institute Animal Care Committee). The mice were housed in cohorts of 5 mice per cage under specific pathogen free conditions.
In first-line treatment experiments, we initiated therapy at an average tumor size of around 200 mm3. In second-line treatment experiments, PC-3 xenograft tumors of 200 mm3 were subjected to metronomic cyclophosphamide chemotherapy until they reached an average size of around 750 mm3, at which point second-line therapy was initiated.
We used the following regimens, either as monotherapies or in combination (n ≥ 5 mice per treatment cohort): (1) metronomic cyclophosphamide: 20 mg/kg/d administered continuously through the drinking water [18]; (2) maximum tolerated dose cyclophosphamide: 100 mg/kg on days 1, 3 and 5 intraperitoneally (21-day treatment cycles) [18]; (3) bolus dose cyclophosphamide: 150 mg/kg given intraperitoneally every 21 days [19]; (4) metronomic docetaxel: 10 mg/kg twice a week intraperitoneally (Man and Kerbel, unpublished observation); (5) maximum tolerated dose docetaxel: 25 mg/kg intraperitoneally every 21 days (as converted from the conventional dose in humans [20]); (6) metronomic oral topotecan: 1 mg/kg by daily gavage [21]; and (7) bevacizumab 100 μg/mouse twice a week intraperitoneally [22]. Of note, for all metronomic regimens we used the optimal biological dose of the respective agent obtained by dose–response analysis of treatment-associated changes of the number of circulating endothelial progenitor cells, as previously reported [23].
cDNA microarray analyses
Ten μg of total RNA obtained from advanced LCR1.1 tumors (~1,500 mm3), and of NS1.1 tumors (n = 3 each) were labeled with cyanine dyes (Cy5 and Cy3)(Amersham Biosciences/GE Health Care Life Sciences, Baie d’Urfe, QC/Canada) and hybridized to Human 19 k v8 cDNA arrays on the Advalytix SlideBooster™ (Advalytix, Germany) using DIG Easy Hyb hybridization solution (Roche Applied Sciences, Indianapolis, IN) at the University Health Network Microarray Centre (http://www.microarrays.ca; Toronto, ON/Canada). The slides were scanned on the Agilent G2565BA scanner (Santa Clara, CA) and quantified using ArrayVision v.8.0 (Imaging Research Inc./GE Health Care Life Sciences). The data and images were then loaded into GeneTraffic™ (Iobion Informatics, Toronto, ON/Canada) for normalization (using the Lowess, sub-grid method) and filtering to remove spots that were flagged, mostly due to signals near background levels. A list of all genes that were not flagged was subjected to one-class Significance Analysis of Microarrays (SAM; version 2.20, Stanford University) [24]. In a second SAM, we only included genes that had absolute values of log2 ratio ≥ 1 in at least 1 observation.
The combined list of differentially regulated genes from both analyses was uploaded and mapped to genes in the Database for Annotation, Visualization and Integrated Discovery (DAVID; v6.7, http://david.abcc.ncifcrf.gov/) [25, 26] for gene enrichment and functional annotation analyses. We applied default settings except that the EASE threshold (p value of a modified Fisher Exact test for gene enrichment) was set at 0.05.
Statistical analyses
Results are reported as mean ± standard deviation unless otherwise indicated. Statistical significance of observed differences was assessed using the PRISM Version 4.00 software (GraphPad, San Diego, CA) and by applying a two-sided level of significance set at p < 0.05.
Results
First-line therapies of established PC-3 tumors: studying intrinsic resistance
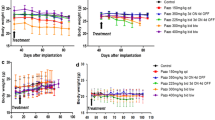
PC-3 human prostate xenograft tumors are exquisitely sensitive to metronomic chemotherapy applying the alkylating agent cyclophosphamide at 20 mg/kg/day given through the drinking water (Fig. 1a), notably a regimen that is very well tolerated (Fig. 1b) and that is of translational relevance [3, 15, 18]. To test treatment sensitivity to other classes of chemotherapeutic agents, first we treated PC-3 tumor bearing mice with metronomic docetaxel monotherapy. While conventional docetaxel is the standard of care first-line chemotherapy in advanced prostate cancer [27], metronomic regimens containing docetaxel have shown activity in a number of phase I/II clinical trials of prostate and other tumor types [28–31]. In addition, metronomic protocols of docetaxel, or of other taxanes, have been successfully applied in breast, gastric and ovarian cancer models [32–35]. In the PC-3 model, metronomic docetaxel resulted in a significant growth delay that was comparable to the effects of metronomic cyclophosphamide, whereas combining docetaxel and cyclophosphamide in one metronomic regimen was not superior to each regimen alone (Fig. 1a). Unexpectedly, mice started to lose weight and became lethargic after 3 weeks of metronomic docetaxel monotherapy (Fig. 1b), thus they had to be sacrificed as per institutional animal care guidelines. The weight loss was even more pronounced in mice receiving combined metronomic docetaxel and cyclophosphamide chemotherapy (Fig. 1b). At necropsy, most metronomic docetaxel treated mice were found to have generalized large bowel distension with signs of fecal impaction (Fig. 2a). The droppings of mice subjected to metronomic docetaxel were not only visibly larger but the average dry weight per dropping was also significantly increased around twofold (Fig. 2b). Of note, we did not observe such a phenomenon in mice treated with maximum tolerated dose docetaxel, with metronomic cyclophosphamide alone, or with a combination of maximum tolerated dose docetaxel plus metronomic cyclophosphamide. Despite a negative impact of maximum tolerated dose docetaxel therapy on mouse weight gain (Fig. 1d), treatment tolerance of conventional docetaxel was superior to metronomic docetaxel, without signs of lethargy or intestinal toxicity. As previously reported, the antitumor effects of maximum tolerated dose docetaxel on PC-3 xenografts were modest (Fig. 1c) [36, 37]. Furthermore, the combination of maximum tolerated dose docetaxel with metronomic cyclophosphamide was not superior to metronomic cyclophosphamide therapy alone (Fig. 1c).

Effects of metronomic versus conventional docetaxel chemotherapy on PC-3 human prostate cancer xenograft tumors: a Low-dose metronomic cyclophosphamide or docetaxel monotherapy (LDM CPA and LDM TAX, respectively) resulted in similar anti-PC-3 effects (both p = 0.006 versus normal saline (NS) control on day 38), which were not further enhanced by combined cyclophosphamide/docetaxel metronomic therapy (p = 0.012 versus NS on day 38). b Mice treated with metronomic docetaxel, and even more so, mice subjected to combined metronomic docetaxel/cyclophosphamide therapy experienced weight loss and became lethargic after 3 weeks of treatment. c Maximum tolerated dose docetaxel was only moderately active in PC-3 tumors (MTD TAX; p = 0.081 versus NS on day 38). Combined MTD TAX and LDM CPA were not more active than LDM CPA alone (p = 0.009 and p = 0.004, respectively, versus NS on day 38). d Despite treatment-associated weight fluctuations, MTD TAX was better tolerated than LDM TAX based on body weight changes over time. * treatment start at day 21, average tumors size of 212 ± 89 mm3

Fecal impaction and intestinal stasis associated with metronomic docetaxel chemotherapy. a After 3 weeks of therapy, mice subjected to metronomic docetaxel became lethargic and started to lose weight. Necropsy revealed intestinal stasis and fecal impaction (arrowhead) in the majority of animals. b The droppings of mice subjected to metronomic docetaxel (LDM TAX) were visibly larger, and their average dry weight was increased more than twofold compared to other treatment cohorts (NS: normal saline; MTD TAX: maximum tolerated dose docetaxel, LDM CPA: metronomic cyclophosphamide). ***, p < 0.001; **, p < 0.01
Next, we decided to test the antitumor activity of oral metronomic topotecan. Successful metronomic applications of this topoisomerase I inhibitor have been described in colorectal, ovarian and pediatric tumor models [21, 38–40]. However, the metronomic oral topotecan regimen we applied failed to show antitumor activity towards established primary PC-3 xenograft tumors (Fig. 3a). Otherwise, we could confirm the excellent tolerance of this treatment protocol (Fig. 3b).

Oral metronomic topotecan therapy does not suppress primary PC-3 tumor growth. a Oral metronomic topotecan (LDM TOPO) does not affect PC-3 tumor growth compared to control gavage (Control). b As suggested by serial weight measurements, LDM TOPO is very well tolerated. * treatment start at day 21, average tumors size of 212 ± 89 mm3
Treatment of PC-3 xenograft tumors resistant to metronomic cyclophosphamide: overcoming acquired resistance
Despite an initial marked response to therapy, PC-3 xenograft tumors subjected to metronomic cyclophosphamide eventually progress [15]. In an attempt to define potential second-line therapies, we decided to switch mice with recurrent PC-3 tumors on metronomic cyclophosphamide therapy to various other cyclophosphamide regimens, to maximum tolerated dose docetaxel, or to bevacizumab therapy. In previous experiments we showed that PC-3 variants with stable, acquired resistance to metronomic cyclophosphamide therapy remained sensitive to maximum tolerated dose cyclophosphamide treatment when the cells were re-implanted into new hosts [15]. Herein, to expand on these findings and to more closely replicate a clinical scenario, we subjected PC-3 tumor bearing mice to metronomic cyclophosphamide therapy until recurrence to an average size of around 750 mm3. Next, we compared the tumor growth pattern of cohorts of mice maintained on metronomic cyclophosphamide versus mice switched either to (i) saline control, or to (ii) maximum tolerated dose cyclophosphamide (300 mg/kg per 21-day cycle), to (iii) a previously described bolus dose cyclophosphamide regimen (150 mg/kg per 21-day cycle) [19], or to (iv) combined bolus dose and metronomic cyclophosphamide (Fig. 4a). Metronomic cyclophosphamide therapy beyond the point of acquired resistance was not significantly superior to normal saline control therapy. In contrast, maximum tolerated dose cyclophosphamide resulted in initial tumor regression followed by delayed progression (Fig. 4a). While the cyclophosphamide bolus dose regimen was less effective than the maximum tolerated dose cyclophosphamide protocol, the combination of bolus dose and metronomic cyclophosphamide resulted in similar tumor growth suppression as the maximum tolerated dose cyclophosphamide regimen (Fig. 4a). All treatments were associated with gradual weight loss coinciding with increasing tumor burden. The weight curves also documented transient weight losses associated with both maximum tolerated dose and bolus dose cyclophosphamide administration (Fig. 4b).

Second-line therapies in PC-3 tumors with in vivo acquired resistance to metronomic cyclophosphamide. a Continuation of low-dose metronomic cyclophosphamide (LDM CPA) beyond progression (i.e., at an average tumor size of around 750 mm3) was not beneficial (p = 0.658 on day 67 compared to normal saline (NS) control). However, compared to NS, bolus dose cyclophosphamide alone (BD CPA), and combined bolus dose and metronomic cyclophosphamide (BD CPA + LDM CPA) delayed the time to an average tumor size of 1500 mm3 by around 14 or 42 days, respectively, even though the degree of initial tumor regression did not reach statistical significance (BD CPA versus NS p = 0.189 on day 67; BD CPA + LDM CPA versus NS p = 0.072 on day 67). Of note, PC-3 tumors with in vivo acquired resistance to LDM CPA remained highly sensitive to maximum tolerated dose cyclophosphamide (MTD CPA versus NS p = 0.037 on day 67). b With increasing tumor and treatment burden there was a trend for progressive weight loss that remained though less than 10 % of the starting weight (i.e., weight at initiation of second-line therapy). c Neither maximum tolerated dose docetaxel (MTD TAX), nor MTD TAX combined with metronomic cyclophosphamide (MTD TAX + LDM CPA), resulted in significant early tumor growth delay (p = 0.429 and 0.196, respectively, versus NS on day 67). However, the time to an average tumor burden per mouse of around 1500 mm3 increased by around 10 days (MTD TAX) or 21 days (MTD TAX and LDM CPA). d As per serial weight measurements, MTD TAX and MTD TAX + LDM CPA were well tolerated. e Treatment with bevacizumab (BEV) or bevacizumab combined with metronomic cyclophosphamide (BEV + LDM CPA) did not produce significant early antitumor effects (p = 0.115 and 0.076, respectively, on day 67), but the time to an average tumor size of around 1500 mm3 was increased by 21 days. BEV + LDM CPA was not superior to BEV monotherapy. f Both bevacizumab treatment regimens were well tolerated
Next, we tested the impact of maximum tolerated dose docetaxel on metronomic cyclophosphamide resistant PC-3 tumors. Similar to the modest antitumor activity seen in treatment-naïve PC-3 tumors (Fig. 1c), maximum tolerated dose docetaxel resulted in a growth delay of a few days only in metronomic cyclophosphamide resistant PC-3 xenografts (Fig. 4c). However, the antitumor effects of maximum tolerated dose docetaxel combined with metronomic cyclophosphamide were superior compared to each treatment alone.
Bevacizumab does not significantly affect the growth of treatment-naïve PC-3 tumors [36]. However, we previously showed that bevacizumab is able to overcome acquired resistance of erbB-2 expressing MDA-MB-231 breast cancer xenograft tumors to combined metronomic cyclophosphamide and trastuzumab (anti-erbB-2 antibody) therapy [41]. Therefore, we decided to study the use of second-line bevacizumab therapy in metronomic cyclophosphamide resistant PC-3 tumors. While bevacizumab monotherapy impaired PC-3 tumor progression, no further enhancement of this antitumor activity was seen with the addition of metronomic cyclophosphamide (Fig. 4e).
cDNA microarray analyses
To obtain molecular insights into the mechanisms of resistance to metronomic cyclophosphamide chemotherapy, we performed cDNA microarray analyses comparing LCR1.1 with NS1.1 tumors [15], as depicted in Fig. 5a. The normalized and filtered expression data was subjected to one-class SAM under two conditions [24]. First, when all genes were included for SAM, 22 were found to be significantly upregulated in LCR1.1 tumors (Supplemental Table 1). None were negatively regulated. Second, we included only genes with absolute values of log2 ratio ≥ 1 in at least one observation for SAM. This analysis revealed 26 upregulated genes, but again no downregulated genes (Supplemental Table 2). These two lists were merged to yield a total of 41 upregulated genes in LCR1.1 tumors (Table 1). Only few of the 41 genes have been clearly associated with cancer in general, or prostate cancer in particular, such as JAK1 and FLI1 [42–45].

cDNA microarray analyses of LCR1.1 versus NS1.1 tumors. a Total RNA of metronomic cyclophosphamide resistant LCR1.1 tumors, and of NS1.1 control tumors were subjected to cDNA microarray analyses followed by Significance Analysis of Microarrays (SAM) and gene enrichment as well as functional annotation analyses. b Neither pro-angiogenic genes (open bars) nor endogenous angiogenesis inhibitors previously implicated in metronomic chemotherapy effects (shaded bars) were differentially regulated (SAM, p > 0.05) in LCR1.1 versus NS1.1 tumors. VEGF A/B: vascular endothelial growth factor A/B; VEGFR1/2: VEGF receptor 1/2; ANG 2: angiopoietin 2; IL-6: interleukin 6; TSP-1: thrombospondin 1; PEDF; pigment epithelium-derived factor
Next, the combined list of genes (Table 1) was imported into the DAVID database for gene enrichment and functional annotation analyses [25, 26]. While the list of upregulated genes in LCR1.1 was significantly enriched for 8 different gene ontology terms, only the term encompassing ‘translation, ribosomal structure and biogenesis’ maintained significance following Bonferroni correction for multiple testing (Table 2). Genes related to translation regulation (i.e., EIF2B1, EIF253, IMP3, PES1) were 17-fold enriched. Interestingly, PES1 contains a BRCT domain commonly found in proteins regulating cell cycle checkpoints following DNA damage, including the breast cancer gene BRCA1 [46]. The BRCT domain feature was around 40-fold enriched in our combined list of upregulated genes. Of note, metronomic cyclophosphamide resistance does not appear to be associated with significant differential changes in pro-angiogenic factors such as vascular endothelial growth factor, or endogenous angiogenesis inhibitors that amplify the antiangiogenic effects of metronomic cyclophosphamide, such as thrombospondin-1 or pigment epithelium-derived factor (Fig. 5b) [47, 48].
Discussion
Since the first use of antifolates and nitrogen mustard in the 1940s, the field of cancer chemotherapy has rapidly evolved with the development and approval of numerous classes of cytotoxic agents with distinct mechanisms of actions, and the capability of safely administering increasingly higher doses of chemotherapeutics alone or in combination [49]. Nonetheless, therapeutic resistance remains one of the major obstacles in systemic cancer therapy to date, and that includes also targeted agents [50, 51]. Furthermore, maximum tolerated dose chemotherapy administration is especially challenging in the elderly and frail due to treatment-associated acute toxic side-effects. In contrast, low-dose metronomic chemotherapy is generally very well tolerated while showing promising antitumor efficacy [2]. However, as is the case with maximum tolerated dose chemotherapy, therapeutic resistance is likely to limit the benefits of metronomic chemotherapy. In addition, there are concerns that metronomic chemotherapy might facilitate the development of resistance to conventional chemotherapy or to antiangiogenic therapies [15].
We recently showed that stable, in vivo acquired resistance to metronomic cyclophosphamide therapy is phenotypically distinct from resistance to maximum tolerated dose cyclophosphamide [15]. In other words, PC-3 human prostate cancer cells that acquired resistance to metronomic cyclophosphamide remained sensitive to maximum tolerated doses of cyclophosphamide. On the other hand, it is important to note that PC-3 tumor variants resistant to maximum tolerated dose cyclophosphamide still responded to metronomic cyclophosphamide, albeit to lesser degrees than parental PC-3 [15].
Herein, we expanded on these previous findings. First, we show that the PC-3 model is not universally responsive to metronomic chemotherapy regimens. While parental PC-3 tumors respond well to metronomic cyclophosphamide and to metronomic docetaxel, they are resistant to metronomic oral topotecan. This contrasts with significant antitumor effects of metronomic topotecan reported in preclinical studies of ovarian and colon cancer, in neuroblastoma, osteosarcoma and rhabdomyosarcoma models [21, 38, 39], and in renal cell cancer (Jedeszko and Kerbel, unpublished observations). Interestingly, Aljuffali et al. recently showed significant anti-PC-3 effects of topotecan given continuously by means of a micro-osmotic pump [52]. Using this subcutaneous method of drug administration, a dose as low as 0.1 mg/kg/day of topotecan enabled PC-3 tumor stabilization over the entire treatment period of 4 weeks. Second, the degree of response to maximum tolerated doses of a given chemotherapeutic agent does not necessarily predict response to the same drug used metronomically. In fact, while PC-3 tumors are exquisitely sensitive both to maximum tolerated dose cyclophosphamide and to low-dose metronomic cyclophosphamide, we observed significant antitumor activity of metronomic docetaxel but only a modest benefit of maximum tolerated dose docetaxel. Third, discontinuation of metronomic cyclophosphamide upon acquired therapeutic resistance does not result in tumor growth acceleration. The latter possibility of ‘rebound’ tumor growth after therapy is stopped has been raised as a concern for patients undergoing targeted antiangiogenic therapies [53, 54]. Fourth, metronomic cyclophosphamide therapy continued beyond the point at which resistance develops is still beneficial in combination with intermittent administration of maximum tolerated dose docetaxel, or bolus dose cyclophosphamide. Finally, the use of metronomic docetaxel resulted in unexpected toxicity, i.e., intestinal stasis and fecal impaction, the nature of which remains to be further investigated. Others have safely administered similar metronomic docetaxel regimens preclinically, albeit using different mouse strains [32–35]. Of note, the cumulative dose of docetaxel administered using the metronomic regimen applied herein (i.e., 60 mg/kg/21 days) was higher than the docetaxel dose of the maximum tolerated dose regimen (i.e., 25 mg/kg/21 days). While the intestinal toxicity seen in our mice could be related to cumulative docetaxel exposure, increasing the dosing frequency while decreasing individual doses at the same time [35], or using implantable continuous-release formulations of docetaxel [55, 56] could be ways to reduce the cumulative dose of docetaxel, possibly without compromising in terms of antitumor effects. However, it is reassuring that intestinal stasis has not been described as a consequence of metronomic docetaxel administration in patients, although intestinal obstruction is considered a very rare clinical side-effect of conventional docetaxel chemotherapy [28–31].
In the absence of robust biomarker guidance, the clinical translation of the metronomic treatment concept is challenging due to the empirical nature by which the metronomic dose and the dosing interval are chosen [2]. Our findings highlight another challenge. Although our studies have been restricted to the PC-3 tumor model and three distinct chemotherapy drugs, the findings suggest that the type of chemotherapy agent chosen for metronomic purposes matters as a function of the tumor to be treated. Furthermore, the therapeutic sensitivity to a given cytotoxic drug used conventionally does not appear to be closely associated with the potential benefit of using the same drug metronomically. This is not entirely unexpected considering the distinct mechanisms of action proposed for metronomic versus conventional chemotherapy [4].
With the exception of its use as monotherapy for adjuvant treatment of early stage micrometastatic disease, metronomic chemotherapy is unlikely to be used alone, especially in situations of rapid tumor progression of advanced stage cancer [5, 6]. However, due to the generally excellent safety profile, metronomic chemotherapy can be added not only safely but also very successfully to other treatment modalities [2, 4]. In this respect, we have shown previously that metronomic cyclophosphamide combined with intermittent bolus cyclophosphamide is superior compared to each treatment alone [19]. In addition, we report here for the first time the same observation for tumors with acquired resistance to metronomic cyclophosphamide monotherapy.
The antiangiogenic activities of metronomic chemotherapy make such regimens an interesting combination partner for targeted antivascular agents such as vascular endothelial growth factor inhibitors [57]. Such combinations are expected to be associated with a lower risk of high-grade adverse events such as myelosuppression and thromboembolic complications, the risk of which is known to increase when antivascular agents are combined with conventional, maximum tolerated dose chemotherapy [58, 59]. Our studies suggest that metronomic chemotherapy and targeted antivascular agents may also be used sequentially. In fact, metronomic cyclophosphamide resistant PC-3 xenografts were sensitive to bevacizumab therapy, even though parental PC-3 tumors do not respond to bevacizumab [36]. These findings are in line with results reported by du Manoir et al. [41]. By studying 231-H2N tumor xenografts (an erbB-2/HER2 expressing MDA-MB-231 breast cancer cell variant) with in vivo acquired resistance to metronomic cyclophosphamide combined with trastuzumab (an anti- erbB-2/HER2 monoclonal antibody), they showed that such resistant tumors were sensitive to adding bevacizumab. On the other hand, in our metronomic cyclophosphamide resistant PC-3 tumors continuing metronomic cyclophosphamide in addition to starting bevacizumab was not superior to bevacizumab alone, even though upfront combinations of metronomic cyclophosphamide and anti-VEGF agents have shown to be beneficial in the PC-3 model [18].
Thus, it appears that despite acquired resistance to metronomic cyclophosphamide monotherapy the continuation of metronomic cylophophosphamide is beneficial when combined with conventional docetaxel or bolus cyclophosphamide administration. Two non-mutually exclusive explanations for these findings might apply. It remains to be seen if metronomic cyclophosphamide is sensitizing tumors to conventional chemotherapy even if these tumors are resistant to metronomic cyclophosphamide monotherapy. Alternatively, conventional chemotherapy may be able to re-sensitize metronomic cyclophosphamide resistant PC-3 tumors to metronomic cyclophosphamide administration. However, such postulated cross-sensitization seems not to take place when metronomic cyclophosphamide is used in conjunction with bevacizumab.
Resistance to antiangiogenic agents targeting the vascular endothelial growth factor pathway is mediated by a number of mechanisms such as: (1) evasive resistance by activation of redundant angiogenic pathways; (2) vascular remodeling resulting in comparatively mature vessels that tend to be less responsive to antiangiogenic treatments compared to newly formed immature capillaries; (3) vascular co-option, the enhanced ability for infiltrative tumor progression into the surrounding normal tissue that depends on the pre-existing vasculature of this normal tissue rather than neoangiogenesis; and (4) reduced vascular dependence, the successful tumor cell adaptation to the hostile microenvironment resulting from long-term antiangiogenic therapy, characterized by oxygen, nutrient and growth factor deprivation [60]. In the PC-3 model resistance to metronomic cyclophosphamide appears to be mediated primarily by reduced vascular dependence [17]. Accordingly, our microarray analyses do not reveal significant expression changes of pro-angiogenic molecules or of endogenous angiogenesis inhibitors in LCR1.1 versus NS1.1 tumors. Although this may be considered inconsistent with the response of metronomic cyclophosphamide resistant PC-3 tumors to bevacizumab, our experimental setup cannot exclude the possibility of increased sensitivity of tumor endothelial cells to vascular endothelial growth factor deprivation in metronomic cyclophosphamide resistant versus treatment-naïve PC-3 tumors.
Microarray analyses of LCR1.1 tumors revealed potentially actionable expression changes such as JAK1 upregulation [43, 61]. In addition, our data indicate that resistance to metronomic cyclophosphamide chemotherapy is associated with altered regulation of protein translation. In comparative proteome analyses, Thoenes et al. identified thioredoxin containing protein 5, cathepsin B and annexin A3 as possible mediators of resistance of PC-3 derived tumors to metronomic cyclophosphamide [16]. None of these genes were found to be differentially regulated in our analyses. Using the same PC-3 models Kubisch et al. performed cDNA microarray analyses that revealed an association of resistance to metronomic cyclophosphamide with three gene ontologies: complement and coagulation cascade, axon guidance and steroid biosynthesis [62]. The lack of overlap in the gene expression changes seen in these two studies compared to our analyses may result from methodological differences. In addition, the cyclophosphamide resistant PC-3 variants used by Thoenes et al. and Kubisch et al. were derived by using a weekly metronomic cyclophosphamide regimen [14], whereas we obtained the LCR1.1 variant by administering cyclophosphamide continuously via the drinking water [18].
In conclusion, as seen with other systemic treatment modalities, metronomic chemotherapy is not devoid of intrinsic or acquired resistance. Herein, we have expanded our previous findings that resistance to metronomic versus conventional chemotherapy is distinct. Altered regulation of protein translation and anti-coagulation amongst others may contribute to resistance to metronomic cyclophosphamide therapy. A better understanding of the molecular mechanisms of such resistance is expected to lead the way to overcome or delay therapeutic resistance to metronomic chemotherapy using cyclophosphamide and possibly also other chemotherapeutic drugs. Such efforts will need to include tumor models other than PC-3.
References
Kerbel RS, Kamen BA (2004) The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer 4(6):423–436
Pasquier E, Kavallaris M, Andre N (2010) Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol 7(8):455–465. doi:10.1038/nrclinonc.2010.82
Nelius T, Rinard K, Filleur S (2011) Oral/metronomic cyclophosphamide-based chemotherapy as option for patients with castration-refractory prostate cancer: review of the literature. Cancer Treat Rev 37(6):444–455. doi:10.1016/j.ctrv.2010.12.006
Emmenegger U, Francia G, Shaked Y, Kerbel RS (2010) Metronomic chemotherapy: principles and lessons learned from applications in the treatment of metastatic prostate cancer. Recent Results Cancer Res 180:165–183. doi:10.1007/978-3-540-78281-0_10
Kato H, Ichinose Y, Ohta M, Hata E, Tsubota N, Tada H, Watanabe Y, Wada H, Tsuboi M, Hamajima N (2004) A randomized trial of adjuvant chemotherapy with uracil-tegafur for adenocarcinoma of the lung. N Engl J Med 350(17):1713–1721
Watanabe T, Sano M, Takashima S, Kitaya T, Tokuda Y, Yoshimoto M, Kohno N, Nakagami K, Iwata H, Shimozuma K, Sonoo H, Tsuda H, Sakamoto G, Ohashi Y (2009) Oral uracil and tegafur compared with classic cyclophosphamide, methotrexate, fluorouracil as postoperative chemotherapy in patients with node-negative, high-risk breast cancer: National Surgical Adjuvant Study for Breast Cancer 01 Trial. J Clin Oncol 27(9):1368–1374
Bottini A, Generali D, Brizzi MP, Fox SB, Bersiga A, Bonardi S, Allevi G, Aguggini S, Bodini G, Milani M, Dionisio R, Bernardi C, Montruccoli A, Bruzzi P, Harris AL, Dogliotti L, Berruti A (2006) Randomized phase II trial of letrozole and letrozole plus low-dose metronomic oral cyclophosphamide as primary systemic treatment in elderly breast cancer patients. J Clin Oncol 24(22):3623–3628
Borne E, Desmedt E, Duhamel A, Mirabel X, Dziwniel V, Maire C, Florin V, Martinot V, Penel N, Vercambre-Darras S, Mortier L (2009) Oral metronomic cyclophosphamide in elderly with metastatic melanoma. Invest New Drugs. doi:10.1007/s10637-009-9298-5
Fontana A, Falcone A, Derosa L, Di Desidero T, Danesi R, Bocci G (2010) Metronomic chemotherapy for metastatic prostate cancer: a ‘young’ concept for old patients? Drugs Aging 27(9):689–696. doi:10.2165/11537480-000000000-00000
Fontana A, Bocci G, Galli L, D’Arcangelo M, Derosa L, Fioravanti A, Orlandi P, Barletta MT, Landi L, Bursi S, Minuti G, Bona E, Grazzini I, Danesi R, Falcone A (2010) Metronomic cyclophosphamide in elderly patients with advanced, castration-resistant prostate cancer. J Am Geriatr Soc 58(5):986–988. doi:10.1111/j.1532-5415.2010.02833.x
Kerbel RS (2012) Strategies for improving the clinical benefit of antiangiogenic drug based therapies for breast cancer. Journal of Mammary Gland Biology and Neoplasia 17(3–4):229–239. doi:10.1007/s10911-012-9266-0
Folkins C, Man S, Xu P, Shaked Y, Hicklin DJ, Kerbel RS (2007) Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res 67(8):3560–3564
Martin-Padura I, Marighetti P, Agliano A, Colombo F, Larzabal L, Redrado M, Bleau AM, Prior C, Bertolini F, Calvo A (2012) Residual dormant cancer stem-cell foci are responsible for tumor relapse after antiangiogenic metronomic therapy in hepatocellular carcinoma xenografts. Lab Invest 92(7):952–966. doi:10.1038/labinvest.2012.65
Browder T, Butterfield CE, Kraling BM, Shi B, Marshall B, O’Reilly MS, Folkman J (2000) Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res 60(7):1878–1886
Emmenegger U, Francia G, Chow A, Shaked Y, Kouri A, Man S, Kerbel RS (2011) Tumors that acquire resistance to low-dose metronomic cyclophosphamide retain sensitivity to maximum tolerated dose cyclophosphamide. Neoplasia 13(1):40–48
Thoenes L, Hoehn M, Kashirin R, Ogris M, Arnold GJ, Wagner E, Guenther M (2010) In vivo chemoresistance of prostate cancer in metronomic cyclophosphamide therapy. J Proteomics 73(7):1342–1354. doi:10.1016/j.jprot.2010.02.019
Emmenegger U, Morton GC, Francia G, Shaked Y, Franco M, Weinerman A, Man S, Kerbel RS (2006) Low-dose metronomic daily cyclophosphamide and weekly tirapazamine: a well-tolerated combination regimen with enhanced efficacy that exploits tumor hypoxia. Cancer Res 66(3):1664–1674
Man S, Bocci G, Francia G, Green SK, Jothy S, Hanahan D, Bohlen P, Hicklin DJ, Bergers G, Kerbel RS (2002) Antitumor effects in mice of low-dose (metronomic) cyclophosphamide administered continuously through the drinking water. Cancer Res 62(10):2731–2735
Shaked Y, Emmenegger U, Francia G, Chen L, Lee CR, Man S, Paraghamian A, Ben-David Y, Kerbel RS (2005) Low-dose metronomic combined with intermittent bolus-dose cyclophosphamide is an effective long-term chemotherapy treatment strategy. Cancer Res 65(16):7045–7051
Graham MA, Senan S, Robin H Jr, Eckhardt N, Lendrem D, Hincks J, Greenslade D, Rampling R, Kaye SB, von Roemeling R, Workman P (1997) Pharmacokinetics of the hypoxic cell cytotoxic agent tirapazamine and its major bioreductive metabolites in mice and humans: retrospective analysis of a pharmacokinetically guided dose-escalation strategy in a phase I trial. Cancer Chemother Pharmacol 40(1):1–10
Hashimoto K, Man S, Xu P, Cruz-Munoz W, Tang T, Kumar R, Kerbel RS (2010) Potent preclinical impact of metronomic low-dose oral topotecan combined with the antiangiogenic drug pazopanib for the treatment of ovarian cancer. Mol Cancer Ther 9(4):996–1006. doi:10.1158/1535-7163.MCT-09-0960
Gerber HP, Ferrara N (2005) Pharmacology and pharmacodynamics of bevacizumab as monotherapy or in combination with cytotoxic therapy in preclinical studies. Cancer Res 65(3):671–680
Shaked Y, Emmenegger U, Man S, Cervi D, Bertolini F, Ben-David Y, Kerbel RS (2005) Optimal biologic dose of metronomic chemotherapy regimens is associated with maximum antiangiogenic activity. Blood 106(9):3058–3061. doi:10.1182/blood-2005-04-1422
Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 98(9):5116–5121. doi:10.1073/pnas.091062498
da Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4(1):44–57. doi:10.1038/nprot.2008.211
da Huang W, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37(1):1–13. doi:10.1093/nar/gkn923
Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Theodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA (2004) Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 351(15):1502–1512
Sanborn SL, Cooney MM, Dowlati A, Brell JM, Krishnamurthi S, Gibbons J, Bokar JA, Nock C, Ness A, Remick SC (2008) Phase I trial of docetaxel and thalidomide: a regimen based on metronomic therapeutic principles. Invest New Drugs 26(4):355–362. doi:10.1007/s10637-008-9137-0
Gorn M, Habermann CR, Anige M, Thom I, Schuch G, Andritzky B, Brandl S, Burkholder I, Edler L, Hossfeld DK, Bokemeyer C, Laack E (2008) A pilot study of docetaxel and trofosfamide as second-line ‘metronomic’ chemotherapy in the treatment of metastatic non-small cell lung cancer (NSCLC). Onkologie 31(4):185–189. doi:10.1159/000118626
Young SD, Lafrenie RM, Clemons MJ (2012) Phase ii trial of a metronomic schedule of docetaxel and capecitabine with concurrent celecoxib in patients with prior anthracycline exposure for metastatic breast cancer. Curr Oncol 19(2):e75–e83. doi:10.3747/co.19.879
Facchini G, Caraglia M, Morabito A, Marra M, Piccirillo MC, Bochicchio AM, Striano S, Marra L, Nasti G, Ferrari E, Leopardo D, Vitale G, Gentilini D, Tortoriello A, Catalano A, Budillon A, Perrone F, Iaffaioli RV (2010) Metronomic administration of zoledronic acid and taxotere combination in castration resistant prostate cancer patients: phase I ZANTE trial. Cancer Biol Ther 10(6):543–548. doi:10.4161/cbt.10.6.12611
Ng SS, Sparreboom A, Shaked Y, Lee C, Man S, Desai N, Soon-Shiong P, Figg WD, Kerbel RS (2006) Influence of formulation vehicle on metronomic taxane chemotherapy: albumin-bound versus cremophor EL-based paclitaxel. Clin Cancer Res 12(14 Pt 1):4331–4338
Wu H, Xin Y, Xiao Y, Zhao J (2012) Low-dose docetaxel combined with (−)-epigallocatechin-3-gallate inhibits angiogenesis and tumor growth in nude mice with gastric cancer xenografts. Cancer Biother Radiopharm 27(3):204–209. doi:10.1089/cbr.2011.1103
Wu H, Xin Y, Zhao J, Sun D, Li W, Hu Y, Wang S (2011) Metronomic docetaxel chemotherapy inhibits angiogenesis and tumor growth in a gastric cancer model. Cancer Chemother Pharmacol 68(4):879–887. doi:10.1007/s00280-011-1563-6
Kamat AA, Kim TJ, Landen CN Jr, Lu C, Han LY, Lin YG, Merritt WM, Thaker PH, Gershenson DM, Bischoff FZ, Heymach JV, Jaffe RB, Coleman RL, Sood AK (2007) Metronomic chemotherapy enhances the efficacy of antivascular therapy in ovarian cancer. Cancer Res 67(1):281–288
Newman SP, Foster PA, Ho YT, Day JM, Raobaikady B, Kasprzyk PG, Leese MP, Potter BV, Reed MJ, Purohit A (2007) The therapeutic potential of a series of orally bioavailable anti-angiogenic microtubule disruptors as therapy for hormone-independent prostate and breast cancers. Br J Cancer 97(12):1673–1682. doi:10.1038/sj.bjc.6604100
Muramaki M, Miyake H, Hara I, Kamidono S (2005) Synergistic inhibition of tumor growth and metastasis by combined treatment with TNP-470 and docetaxel in a human prostate cancer PC-3 model. Int J Oncol 26(3):623–628
Hackl C, Man S, Francia G, Milsom C, Xu P, Kerbel RS (2012) Metronomic oral topotecan prolongs survival and reduces liver metastasis in improved preclinical orthotopic and adjuvant therapy colon cancer models. Gut. doi:10.1136/gutjnl-2011-301585
Merritt WM, Danes CG, Shahzad MM, Lin YG, Kamat AA, Han LY, Spannuth WA, Nick AM, Mangala LS, Stone RL, Kim HS, Gershenson DM, Jaffe RB, Coleman RL, Chandra J, Sood AK (2009) Anti-angiogenic properties of metronomic topotecan in ovarian carcinoma. Cancer Biol Ther 8(16):1596–1603
Kumar S, Mokhtari RB, Sheikh R, Wu B, Zhang L, Xu P, Man S, Oliveira ID, Yeger H, Kerbel RS, Baruchel S (2011) Metronomic oral topotecan with pazopanib is an active antiangiogenic regimen in mouse models of aggressive pediatric solid tumor. Clin Cancer Res 17(17):5656–5667. doi:10.1158/1078-0432.CCR-11-0078
du Manoir JM, Francia G, Man S, Mossoba M, Medin JA, Viloria-Petit A, Hicklin DJ, Emmenegger U, Kerbel RS (2006) Strategies for delaying or treating in vivo acquired resistance to trastuzumab in human breast cancer xenografts. Clin Cancer Res 12(3 Pt 1):904–916
Tam L, McGlynn LM, Traynor P, Mukherjee R, Bartlett JM, Edwards J (2007) Expression levels of the JAK/STAT pathway in the transition from hormone-sensitive to hormone-refractory prostate cancer. Br J Cancer 97(3):378–383. doi:10.1038/sj.bjc.6603871
Chang YM, Kung HJ, Evans CP (2007) Nonreceptor tyrosine kinases in prostate cancer. Neoplasia 9(2):90–100
Paulo P, Barros-Silva JD, Ribeiro FR, Ramalho-Carvalho J, Jeronimo C, Henrique R, Lind GE, Skotheim RI, Lothe RA, Teixeira MR (2012) FLI1 is a novel ETS transcription factor involved in gene fusions in prostate cancer. Genes Chromosomes Cancer 51(3):240–249. doi:10.1002/gcc.20948
Erkizan HV, Uversky VN, Toretsky JA (2010) Oncogenic partnerships: EWS-FLI1 protein interactions initiate key pathways of Ewing’s sarcoma. Clin Cancer Res 16(16):4077–4083. doi:10.1158/1078-0432.CCR-09-2261
di Masi A, Gullotta F, Cappadonna V, Leboffe L, Ascenzi P (2011) Cancer predisposing mutations in BRCT domains. IUBMB life 63(7):503–512. doi:10.1002/iub.472
Bocci G, Francia G, Man S, Lawler J, Kerbel RS (2003) Thrombospondin 1, a mediator of the antiangiogenic effects of low-dose metronomic chemotherapy. Proc Natl Acad Sci U S A 100(22):12917–12922
Jia L, Waxman DJ (2013) Thrombospondin-1 and pigment epithelium-derived factor enhance responsiveness of KM12 colon tumor to metronomic cyclophosphamide but have disparate effects on tumor metastasis. Cancer Lett 330(2):241–249. doi:10.1016/j.canlet.2012.11.055
Chabner BA, Roberts TG Jr (2005) Timeline: chemotherapy and the war on cancer. Nat Rev Cancer 5(1):65–72. doi:10.1038/nrc1529
Raguz S, Yague E (2008) Resistance to chemotherapy: new treatments and novel insights into an old problem. Br J Cancer 99(3):387–391. doi:10.1038/sj.bjc.6604510
Bock C, Lengauer T (2012) Managing drug resistance in cancer: lessons from HIV therapy. Nat Rev Cancer 12(7):494–501. doi:10.1038/nrc3297
Aljuffali IA, Mock JN, Costyn LJ, Nguyen H, Nagy T, Cummings BS, Arnold RD (2011) Enhanced antitumor activity of low-dose continuous administration schedules of topotecan in prostate cancer. Cancer Biol Ther 12(5):407–420
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS (2009) Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15(3):232–239. doi:10.1016/j.ccr.2009.01.021
Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O (2009) Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15(3):220–231
De Souza R, Zahedi P, Badame RM, Allen C, Piquette-Miller M (2011) Chemotherapy dosing schedule influences drug resistance development in ovarian cancer. Mol Cancer Ther 10(7):1289–1299. doi:10.1158/1535-7163.MCT-11-0058
De Souza R, Zahedi P, Moriyama EH, Allen CJ, Wilson BC, Piquette-Miller M (2010) Continuous docetaxel chemotherapy improves therapeutic efficacy in murine models of ovarian cancer. Mol Cancer Ther 9(6):1820–1830. doi:10.1158/1535-7163.MCT-10-0249
Klement G, Baruchel S, Rak J, Man S, Clark K, Hicklin DJ, Bohlen P, Kerbel RS (2000) Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest 105(8):R15–R24
Ma L, Francia G, Viloria-Petit A, Hicklin DJ, du Manoir J, Rak J, Kerbel RS (2005) In vitro procoagulant activity induced in endothelial cells by chemotherapy and antiangiogenic drug combinations: modulation by lower-dose chemotherapy. Cancer Res 65(12):5365–5373
Ranpura V, Hapani S, Wu S (2011) Treatment-related mortality with bevacizumab in cancer patients: a meta-analysis. Jama 305(5):487–494. doi:10.1001/jama.2011.51
Ebos JM, Lee CR, Kerbel RS (2009) Tumor and host-mediated pathways of resistance and disease progression in response to antiangiogenic therapy. Clin Cancer Res 15(16):5020–5025. doi:10.1158/1078-0432.CCR-09-0095
Verstovsek S (2013) Ruxolitinib: an oral Janus kinase 1 and Janus kinase 2 inhibitor in the management of myelofibrosis. Postgraduate medicine 125(1):128–135. doi:10.3810/pgm.2013.01.2628
Kubisch R, Meissner L, Krebs S, Blum H, Gunther M, Roidl A, Wagner E (2013) A comprehensive gene expression analysis of resistance formation upon metronomic cyclophosphamide therapy. Transl Oncol 6(1):1–9
Acknowledgements
These studies were conducted with the support from the Ontario Institute for Cancer Research through funding provided by the Government of Ontario as well as by a Prostate Cancer Canada Clinician-Scientist Award to U. Emmenegger, and by grants to Robert S. Kerbel from the Canadian Institutes for Health Research (CIHR) and the National Institutes of Health (CA-41233), USA. Robert S. Kerbel is a Tier I Canada Research Chair in Tumor Biology, Angiogenesis, and Antiangiogenic Therapy. G. Francia was in part supported by University of Texas at El Paso URI and IDR2 grants. We thank Cynthia M. Rodriguez and Karla Parra for their help with the preparation of this manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
One-Class SAM Analysis (all genes) (DOC 41 kb)
Supplementary Table 2
SAM Analysis (genes with absolute values of log2 ratio ≥ 1 in at least one observation) (DOC 74 kb)
Rights and permissions
About this article
Cite this article
Chow, A., Wong, A., Francia, G. et al. Preclinical analysis of resistance and cross-resistance to low-dose metronomic chemotherapy. Invest New Drugs 32, 47–59 (2014). https://doi.org/10.1007/s10637-013-9974-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-013-9974-3