
Abstract
Purpose
To report the clinical and electrophysiological features of cone dystrophy with supernormal rod response (CDSRR).
Methods
Retrospective cohort study of 15 unrelated patients (nine males and six females, median age 16, range 5–47 years) diagnosed with CDSRR by clinical examination, full-field electroretinography (ERG) and genetic testing.
Observations
History, ophthalmic examination including near vision, color vision and contrast sensitivity assessment, multimodal retinal imaging and ERG. Genetic testing was done for all patients using next-generation sequencing.
Results
The rate of consanguinity was 86.7%. Color vision was defective in 56.3%. Near vision was defective in all patients (mean 20/160). Contrast sensitivity was affected in all patients at low contrast of 2.5%. A parafoveal ring of increased autofluorescence imaging was seen in most patients (75%). Supernormal mixed maximal response b-wave was seen bilaterally in 63% of patients (and high normal in 37%). Rod dysfunction with prolonged rod b-wave latency was detected in all. The 30-Hz flicker response was more reduced and delayed compared to the single-flash cone response. A novel homozygous missense variant c.530G>C (p.Cys177Ser) in KCNV2 was detected in one patient, the nonsense homozygous mutation c.427G>T (p.Glu143*) was found in 13 patients, and the nonsense c.159C>G (p.Tyr53*) was found in one patient.
Conclusion
This is the largest cohort of CDSRR from a single ethnic background. Rod dysfunction and reduced 30-Hz flicker response were demonstrated in all patients. In contrast to previous descriptions in the literature, a supernormal combined dark-adapted rod-cone ERG was present in the majority of the patients at standard stimulus intensity. Considering the consistent genotype and the demonstration of likely pathogenic genetic variants in all the patients, we argue that the combination of delayed rod b-wave and subnormal flicker response strongly suggests the diagnosis of CDSRR.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cone dystrophy with supernormal rod responses (CDSRRs) is a rare form of cone dystrophy that was first described by Gouras et al. in 1983 [1]. However, the disease may be more common in populations with a high prevalence of consanguineous marriage. CDSRR is characterized by dysfunction of the cone system associated with reduced sensitivity of rods to dim light and exaggerated rod response to high-intensity stimulus. Patients affected by this condition typically present in the first two decades of life suffering from reduced visual acuity (VA), photophobia, reduced color perception and, in some, in addition nyctalopia. The fundal appearance could be normal in the early stages of the disease, and it may show macular changes later on [2].
The disease (MIM: 610356) has been shown to be associated with recessive mutations in potassium voltage-gated channel subfamily V member 2 (KCNV2) gene (OMIM: 607604) on chromosome 9 (9p24.2) [3, 4]. The KCNV2 gene encodes for the subunit (Kv8.2) of the voltage-gated potassium channels expressed in rods and cones [3]. KCNV2 messenger RNA has been identified in human photoreceptor inner segments, consistent with dysfunction that occurs after phototransduction but before the generation of the full-field electroretinographic b-wave, which is considered to be driven by the rod ON bipolar cells and Muller cells [3]. CDSRR is thought to be the first disorder related to potassium channel dysfunction to affect the visual pathway in humans. CDSRR is believed to be a monogenic disease linked only to KCNV2 mutations [4].
Electroretinogram (ERG) is a very useful tool to diagnose CDSRR with pathognomonic features including reduced and delayed cone responses in addition to reduced rod-specific response and supernormal b-wave of the mixed maximal response on increasing the intensity of the light stimulus [5,6,7].
Previous publications reported features of CDSRR in different ethnic groups [1,2,3,4,5,6,7]. To the best of our knowledge, only one previous publication reported ocular motility disorders associated with this disease in Arab children [8]. In this study, we characterize phenotypic and genotypic features of CDSRR among a cohort of affected patients from a single ethnic background with a high prevalence of consanguineous marriage. We hypothesize that the phenotype may be more consistent and potentially more distinctive within such a population.
Materials (subjects) and methods
This is an observational retrospective cohort study. Institutional Review Board (IRB)/Ethics Committee approval at King Khaled Eye Specialist Hospital (KKESH) was obtained (RP-1751). Informed consent for inclusion in the study was obtained from affected family members or their guardians. Consent for genetic testing was obtained for all cases. The study adheres to the tenets of the Declaration of Helsinki.
We identified 15 patients from 15 families with clinical and electrophysiological diagnosis of CDSRR.
Color fundus photographs were obtained using Topcon fundus camera (Top TRC-50DX) and wide field imaging (Optos PLC, Dunfermline, UK). Fundus autofluorescence (AF) imaging was done using wide field Optos system (Optos 200TX) with 488 nm wavelength.
Contrast sensitivity was tested using Lea Symbol chart held in light box at 3 Microsoft distance with even illumination of 70 Lux. If top symbol was not visible at 3 m distance, the test was repeated at 1.5 m. Presenting uncorrected visual acuity (UCVA) and best-corrected visual acuity (BCVA) was recorded for each eye in LogMAR notation using charts of different contrast sensitivity 100%, 25% and 2.5%. Near vision was recorded by Lea Symbols near chart at 40 cm distance.
The color vision test was done binocularly by quantitative Panel D-15 test. Testing was done in normal daylight. We placed all 15 caps of color in front of patient. We showed the first cap titled pilot and asked the participant to pick near matching caps. Once this was done, pilot was kept in the glass container. This process was repeated till all caps were selected and placed in the glass container.
Retinal structure was analyzed qualitatively with 6 mm transfoveal horizontal spectral-domain optical coherence tomography scans (OCT, Heidelberg Engineering, Inc., Heidelberg, Germany). In addition, the Spectralis macular raster consisting of 19 horizontal 6 mm lines scans was performed.
Retinal function was evaluated with full-field electroretinography (ffERG, Nicolet Biomedical Instruments, Madison, Wisconsin, USA), in dark-adapted and light-adapted state according to ISCEV standards [9], with a few modifications as follows. Full-field electroretinograms were recorded in a Nicolet analysis system (Nicolet Biomedical Instruments, Madison, Wisconsin, USA), after dark adaptation of subjects for 40 min, dilatation of the pupils with topical cyclopentolate 1% and metaoxedrine (phenylephrine hydrochloride) 2.5% and topical anesthesia, with a Burian–Allen bipolar contact lens and a ground electrode applied to the forehead. Responses were obtained stimulating with single full-field flash (30 ms) with blue light (0.81 cd s/m2: rod response) and with white light (10.02 cd s/m2: combined rod-cone response). During ffERG, we used 0.81 cd s/m2 dim blue light for rod response measurement. It is supposedly brighter than the suggested ISCEV standard 0.01 cd s/m2 white flash; however, it does produce an isolated rod response similar to the one suggested ISCEV standard. We have more than 30 years experience using this stimulus, and it did never produce any significant a-wave (such as that seen in the combined rod–cone response), in any patients or normal controls, which indicates that the flash is not excessively bright. Photopic responses were obtained with a background illumination of 3.4 cd s/m2 in order to saturate the rods, after a period of 10 min of light adaptation using the same background illumination.
Molecular biology methods Molecular genetic testing was done for all patients by Center for Human Genetics Bioscientia, Ingelheim, Germany. The two exons and exon–intron boundaries of the KCNV2 gene on chromosome 9p24.2 (OMIM: *607604) were amplified by polymerase chain reaction (PCR) and sequenced directly. The resulting data were compared with the reference sequence NM_133497.3. In patient 4, segregation analysis was done for the parents of the patient as well.
Data analysis The data were entered using Microsoft XL®. The frequency and percentage proportions were calculated. For quantitative variables, nomogram was plotted and mean and standard deviation (SD) values were presented if distribution was normal. In patient of skewed deviation, we calculated median and interquartile range.
Results
The demographics and ocular profile are summarized in Table 1. The age ranged from 5 to 47 year (median: 15.5, IQR: 10). Parental consanguinity was common (13/15 families, 86.7%). In all these consanguineous families, parents were first cousins. Only one family had two affected boys, while the rest had only one affected child. Nystagmus was reported in 9/15 patients (60%). It was noted early in life and improved over time in all affected nine patients. Myopia was found in all 15 cases, and spherical equivalent ranged between − 0.5D and − 11.0D (median: − 5.75D, 25% quartile: − 6.9). Photophobia was evident in 10/15 patients (66.7%). Nyctalopia was reported in 11/15 patients (73.3%).
Visual functions assessment is summarized in Table 2. Distance VA ranged between 20/40 and 20/300 (median: 20/200). Near VA was defective in all patients, ranging between 0.1 (20/25) and 1.12 (20/320) with a mean of 0.9 (20/160). Color vision testing was normal in 7/15 patients (46.7%), mild-moderately defective in 6/15 patients (40%) and severely defective in 2/15 patients (13.3%). Among those with defective color vision, deutran defect was seen in four patients (25%), tritan defect was seen in two patients (12.5%), and a mixture of all axes was seen in two patients (12.5%). Contrast sensitivity was defective in all but one patient (93.3%) with low contrast level of 2.5%.
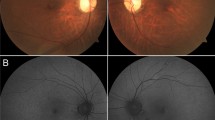
Fundus examination revealed visible choroidal vasculature around the optic disk (especially nasal to the disk) in all patients (100%) with some variability regarding the prominence of this feature, as shown in Fig. 1a, b. The foveal appearance was normal in five patients (33.3%). This is shown in Fig. 1c, d. Slight retinal pigment epithelium (RPE) mottling at the fovea was seen in seven patients (46.7%) as shown in Fig. 1e, f. Central foveal atrophy was seen in four patients (26.7%) illustrated in Fig. 1g, h.

Color fundus photographs in a cohort of 15 patients affected by cone dystrophy with supernormal rod response. a and b Right and left fundus of patient 1 with nonsense homozygous variant c.427G>T (p.Glu143*). Note the choroidal vasculature appearance around the disks. c and d Right and left fundus of patient 13 with homozygous missense variant c.530G>C (p.Cys177Ser) showing normal macular appearance. e and f Right and left fundus of patient 7 with nonsense homozygous variant c.427G>T (p.Glu143*), showing mild foveal retinal pigment epithelium mottling. g and h Right and left fundus of patient 2 with nonsense homozygous variant c.427G>T (p.Glu143*), showing central foveal atrophy
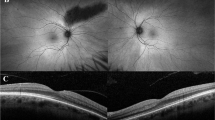
The most prevalent appearance on AF imaging was a parafoveal ring of increased AF signal. This was seen in 13/15 patients (86.7%). Figure 2a, b shows this appearance in a patient with a normal-looking fundus. Central foveal increased AF was seen only in patient 2 (6.6%) shown in Fig. 2c, d. Foci of increased AF signal in the central foveal region (that normally exhibit low AF signal) were seen in patient 8 (Fig. 2e, f). Central macular atrophy (black appearance in AF) surrounded by a ring of increased AF was seen in four patients (26.6%) (patients 3, 6, 7 and 15) illustrated in Fig. 2g, h.

Fundus autofluorescence (AF) images in a cohort of 15 patients affected by cone dystrophy with supernormal rod response. a and b Right and left AF imaging of patient 13 with homozygous missense variant c.530G>C (p.Cys177Ser) showing a parafoveal ring of increased AF. Note the normal fundus appearance of this patient in Fig. 1c and d. c and d Right and left AF imaging of patient 1 with nonsense homozygous variant c.427G>T (p.Glu143*), showing increased central foveal AF. e and f Right and left AF imaging of patient 7 with nonsense homozygous variant c.427G>T (p.Glu143*), showing foci of increased AF in the central foveal area (of normal reduced AF signal). g and h Right and left AF imaging in patient of patient 2 with nonsense homozygous variant c.427G>T (p.Glu143*), showing central foveal atrophy surrounded by a ring of increased AF signal
OCT imaging was unremarkable in one patient (6.6%) highlighted in Fig. 3a, b. In the rest of patients, it revealed three patterns of outer retinal abnormalities. The most common pattern was minimal disruption of inner segment ellipsoid zone (ISe) in seven patients (46.7%), which is illustrated in Fig. 3c, d. Loss of ISe in the central fovea was seen in two patients (13.3%) which is shown in Fig. 3e, f. Loss of ISe and RPE with increased visibility of choroid in Fig. 3g, h was seen in five patients (33.3%).

Transfoveal spectral-domain optical coherence tomography (SD-OCT) imaging in a cohort of 15 patients affected by cone dystrophy with supernormal rod response. a and b Right and left OCT imaging of patient 13 with homozygous missense variant c.530G>C (p.Cys177Ser) showing intact outer retinal layers. c and d Right and left OCT imaging of patient 2 with nonsense homozygous variant c.427G>T (p.Glu143*), showing minimal disruption of inner segment ellipsoid (ISe).e and f Right and left OCT imaging of patient 3 with nonsense homozygous variant c.427G>T (p.Glu143*), showing loss of ISe at the central foveal region. g and h Right and left OCT imaging of patient 6 with nonsense homozygous variant c.427G>T (p.Glu143*), showing loss of ISe and retinal pigment epithelium (RPE) at the central foveal region (with increased visibility of choriocapillaries)
FfERG testing results are summarized in supplement 1 and illustrated in Fig. 4. The rod-specific response was non-recordable in ten eyes from five patients (33.3%). In the other ten patients, the amplitude of rod b-wave was normal in eight (53.3%) patients and subnormal in two patients (13.3%). In all these ten patients (100%), the implicit time of rod b-wave was prolonged beyond normal range. The mixed maximal (rod–cone) response a-wave amplitude was normal in 25 eyes (83.3%) and subnormal in five eyes (15.6%). However, the implicit time of that a-wave was prolonged beyond normal range in all eyes (100%). The mixed maximal response b-wave amplitude was supernormal (above 705 µv) in 20 eyes (66.6%), and it was within the high normal range in the remaining ten eyes (33.3%). The mixed maximal response b-wave implicit time was normal in 20 eyes (66.6%) and slightly prolonged in ten eyes (33.3%). The cone a-wave amplitude was above the normal limit in 15 eyes (50%), and it was within normal range in 15 eyes (50%). The cone a-wave implicit time was prolonged beyond the normal limit in all 30 eyes (100%). The cone b-wave amplitude was subnormal in seven eyes (23.3%) and normal in the remaining 23 eyes (76.6%). The cone b-wave implicit time was prolonged in 19 eyes (63.3%) and normal in 11 eyes (36.6%). The flicker response was non-recordable in four eyes from two patients (13.3%). The flicker b-wave amplitude was subnormal in all but one of the remaining patients (where it just came above the normal lower limit). The flicker b-wave implicit time was prolonged in all the 13 patients where it was recordable. Figure 5 illustrates the ffERG in patient 9 with typical appearance noticed in most of our patients.

Full-field electroretinography amplitude and latency data in a cohort of 15 patients affected by cone dystrophy with supernormal rod response. a Amplitude values in uV compared to normal. b Latency values in ms compared to normals

Full-field electroretinogram recordings in 7-year-old male patient affected by CDSRR with homozygous missense variant c.530G>C (p.Cys177Ser) representing the cohort of patients. The scale is 100 uV, 20 ms. OD = right eye. OS = left eye. Note the normal rod-specific b-wave amplitude with prolonged latency in the left eye. The mixed (rod–cone) response reveals a supernormal b-wave. The cone response shows subnormal b-wave amplitude (almost appears electronegative) and prolonged latency. The flicker response is severely diminished compared to normal
Molecular genetic testing of the patients revealed three variants in KCNV2 gene (Table 3). In patient 13, it revealed the missense variant c.530G>C (p.Cys177Ser) in exon 1 in a homozygous state. This variant has not been described previously in the literature, and it was not found in gnomAD (https://gnomad.broadinstitute.org/, access date 20191228). To the best of our knowledge, this variant has not been annotated in databases, although a similar variant (c.529T>C) affecting the same amino acid position but changing Cysteine to Arginine (p. Cys177Arg) has been described in the literature in Japanese population [10]. However, segregation analysis was not possible in this family. In patient 4, the nonsense mutation c.159C>G (p.Tyr53*) was detected. This variant has already been described in CDSRR [8]. In the remaining 13 patients, the nonsense mutation c.427G>T (p.Glu143*) was detected in exon 1 in a homozygous state. This variant has already been described in the literature [4, 8].
Discussion
In this study, we present the clinical and electrophysiological features of 15 patients (from 15 families, comprising 13 consanguineous marriages) affected by CDSRR with genetic testing in all patients. Robson et al. presented features of this disease in 24 patients from different ethnic backgrounds [11]. To our knowledge, this is the largest series of patients from a similar ethnic group.
The visibility of the choroidal vasculature nasal to the disk that was noted in all patients in this study may be a feature of myopia or CDSRR or both. FfERG testing revealed absent rod-specific response in five patients and delayed response in the remaining 10 patients (with reduced amplitude in three of them). Nyctalopia was a prominent symptom in 13 patients (86.7%). Both of these features give an indication that this disorder considerably affects the rod system function. The photophobia reported in 10/15 patients (66.7%), the reduced near vision in all patients (100%), reduced color vision in 8/14 patients (57%) and reduced contrast sensitivity at low contrast (2.5) in 100% of patients in whom it was tested (n = 14) confirm reduced macular (cone system) function.
In this cohort, the mixed maximal (rod–cone) response (b-wave) was supernormal in 63% of patients and high normal range in 37%. It has been reported that the mixed scotopic b-wave could be in the high normal range in patients of CDSRR [8]. However, in this study, we elicited the supernormal response without increasing the stimulus intensity above the standard settings. This is in contrast to Robson et al. study [11], where they needed to increase the stimulus intensity to obtain such responses. The “square shaped” a-wave in the rod–cone response on exposure to high-intensity stimulus has been suggested as a specific ERG sign for CDSRR [7, 11]. In our cohort, the combination of delayed rod b-wave, delayed rod–cone a-wave, delayed cone a-wave and subnormal flicker response, at standard stimulus intensity, was seen in all patients. This is in contrast to retinitis pigmentosa where typically there is no recordable rod response in combination with additional clinical features such as peripheral bone spicules and vascular attenuation, features that were not seen in this cohort. However, five out of 15 patients in this study did not have any recordable rod-specific response and the remaining had a delayed rod response. Thus, some variability was observed in the electroretinographic features of this cohort. On the other hand, all presented with either a supernormal maximal (rod–cone) response or a response in the upper range of normal for this parameter, combined with a reduced (in all but 1) and delayed 30 Hz flicker cone response, thus indicating a specific combination of rod and cone dysfunction compatible with CDSRR. No particular factors could be observed to be associated with the variable appearance of mixed maximal (rod–cone) response in our cohort. Given the demonstration of pathogenic variants in all 15 patients in KCNV2, we argue that the combination of these ERG features implies a diagnosis of CDSRR.
AF imaging helped to highlight abnormal distribution of fluorophores even in the fundi where the fovea appeared normal clinically and when OCT showed normal outer retina. Parafoveal ring of increased AF was the most commonly seen pattern. This highlights the importance of AF in diagnosis of CDSRR. More atrophic changes on AF and more outer retinal loss were noted in older patients compared to younger ones.
The most common variant among our cohort of patients with genetic testing was nonsense homozygous c.427G>T (p.Glu143*) which has already been described in the literature [4]. One additional novel variant was seen in one patient (missense variant c.530G>C (p.Cys177Ser) in exon 1 in a homozygous state.
Limitations of this study include lack of further tests of macular function such as pattern ERG (PERG) or multifocal ERG (mfERG), lack of visual fields assessment and dark adaptometry and lack of segregation analysis of the novel variant in KCNV2. Furthermore, we were not able to assess patients over longitudinal follow-up. The strength of the study is the detailed full-field electroretinographic assessment of visual function in the largest presented, clinically consistent cohort from a single ethnic background, which may give the opportunity to discover novel distinctive phenotypic features. In contrast to previous descriptions in the literature, a supernormal combined dark-adapted rod–cone ERG was present in the majority of the patients already at standard stimulus intensity. Considering the consistent genotype, we argue that the combination of delayed rod b-wave, delayed rod–cone a-wave, delayed cone a-wave and subnormal 30 Hz flicker response point to the diagnosis of CDSRR. Abnormalities in the a-wave of the rod and cone systems in our cohort suggest lack of function of the potassium channel in addition leads to a defective depolarization of the photoreceptors.
References
Gouras P, Eggers HM, MacKay CJ (1983) Cone dystrophy, nyctalopia, and supernormal rod responses. A new retinal degeneration. Arch Ophthalmol 101(5):718–724
Michaelides M, Holder GE, Webster AR, Hunt DM, Bird AC, Fitzke FW et al (2005) A detailed phenotypic study of “cone dystrophy with supernormal rod ERG”. Br J Ophthalmol 89(3):332–339
Wu H, Cowing JA, Michaelides M, Wilkie SE, Jeffery G, Jenkins SA et al (2006) Mutations in the gene KCNV2 encoding a voltage-gated potassium channel subunit cause “cone dystrophy with supernormal rod electroretinogram” in humans. Am J Hum Genet 79(3):574–579
Wissinger B, Dangel S, Jagle H, Hansen L, Baumann B, Rudolph G et al (2008) Cone dystrophy with supernormal rod response is strictly associated with mutations in KCNV2. Invest Ophthalmol Vis Sci 49(2):751–757
Alexander KR, Fishman GA (1984) Supernormal scotopic ERG in cone dystrophy. Br J Ophthalmol 68(2):69–78
Stockman A, Henning GB, Michaelides M, Moore AT, Webster AR, Cammack J et al (2014) Cone dystrophy with “supernormal” rod ERG: psychophysical testing shows comparable rod and cone temporal sensitivity losses with no gain in rod function. Investig Ophthalmol Vis Sci 55(2):832–840
Vincent A, Robson AG, Holder GE (2013) Pathognomonic (diagnostic) ERGs. A review and update. Retina 33(1):5–12
Khan AO, Alrashed M, Alkuraya FS (2012) ‘Cone dystrophy with supranormal rod response’ in children. Br J Ophthalmol 96(3):422–426
McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R et al (2015) ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol 131(1):81–83
Fujinami K, Tsunoda K, Nakamura N, Kato Y, Noda T, Shinoda K et al (2013) Molecular characteristics of four Japanese cases with KCNV2 retinopathy: report of novel disease-causing variants. Mol Vis 19:1580–1590
Robson AG, Webster AR, Michaelides M, Downes SM, Cowing JA, Hunt DM et al (2010) Cone dystrophy with supernormal rod electroretinogram: a comprehensive genotype/phenotype study including fundus autofluorescence and extensive electrophysiology. Retina 30(1):51–62
Funding
No funding was received for this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers’ bureaus; membership, employment, consultancies, stock ownership or other equity interest; and expert testimony or patent-licensing arrangements), or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
Statement of human rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of the KKESH IRB and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Statement on the welfare of animals
No animals were involved in this research.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Abdelkader, E., Yasir, Z.H., Khan, A.M. et al. Analysis of retinal structure and function in cone dystrophy with supernormal rod response. Doc Ophthalmol 141, 23–32 (2020). https://doi.org/10.1007/s10633-020-09748-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10633-020-09748-1