
Abstract
Purpose
To describe cases of unilateral cone–rod dysfunction presenting in two middle-aged females.
Methods
This case series highlights two middle-aged female patients with progressive visual decline in one eye. Fundus photography, fundus autofluorescence (FAF), spectral-domain optical coherence tomography (SD-OCT), multi-focal electroretinogram (mfERG), full-field electroretinogram(ffERG), and genetic testing were obtained.
Results
In the first patient, mfERG showed an extinguished response and ffERG demonstrated markedly reduced a-wave and b-wave amplitudes (more pronounced under photopic conditions) in the right eye. SD-OCT showed attenuation of the ellipsoid zone of the right eye. Similar findings were appreciated in the second patient. Genetic testing in the first patient identified three heterozygous variants in PRPH2, RCBTB1, and USH2A. The second patient was found to have heterozygous variants in BBS1 and ABCA4.
Conclusion
These two cases add to the literature of case reports of unilateral cone–rod and rod-cone dystrophies. However, the underlying etiology of the unilateral pattern of cone–rod dysfunction and the significance of the heterozygous mutations found in both cases remains uncertain.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ophthalmologists have wrestled with the diagnosis of slowly progressive monocular visual loss for many decades (Francois and Verriest 1952, Franceschetti, Francois, and Babel, 1963) [1, 2]. The gradual loss of cone and/or rod function in one eye can occur from acquired causes such as trauma, medication toxicity (i.e., phenothiazine, chloroquine, hydroxychloroquine), infection (i.e., syphilis, rubella, toxoplasmosis), and inflammation. The presentation of unilateral cone and/or rod dysfunction may also be a highly asymmetric manifestation of a bilateral hereditary disease.
In recent decades, the rapid increases in our understanding of genetic disease have reinforced the notion that nearly all hereditary retinal conditions are bilateral due to germ-line mutations in genes critical for ocular development and function. As both eyes in any one patient have the same parents, it is problematic to fit monocular cases into this framework. The term “dystrophy” has even acquired a connotation of implied underlying genetic etiology, although this is certainly not intrinsic to the etymology of the word which is the Greek equivalent of “bad nutrition or growth.”
Case reports consistently turn up of unilateral cone–rod dystrophies or rod-cone dystrophies (i.e., retinitis pigmentosa) in which acquired etiologies have been ruled out and there are truly no signs of disease in the fellow eye [3,4,5,6,7]. Recently with the advent of genetic testing, there been cases describing unilateral phenotypes with an associated germ-line mutation [8,9,10]. An intriguing example of a patient with a small patch of apparent retinitis pigmentosa-like retinopathy in one eye is shown in Fig. 1. This patient exhibits a localized region of typical bone-spicule pigmentation (Fig. 1A, B) with normal OCT and a corresponding temporal visual field defect in the right eye (Fig. 1D). The patient belongs to a three-generation pedigree of family members with retinitis pigmentosa. Both the patient and her father have documented heterozygous mutations in the rhodopsin gene RHO, which typically causes retinitis pigmentosa as an autosomal dominant trait.

Fundus photos A and autofluorescence B show localized pigmentary retinopathy of the right eye. SD-OCT of the macula C appears normal. Goldmann visual fields C show a inferotemporal visual field defect of the right eye corresponding to the fundus appearance. There is a family history of retinitis pigmentosa, apparently inherited in autosomal dominant fashion. Both the patient and her father carry the same pathogenic mutation in the rhodopsin gene, RHO c.511C > T, p. (Pro171Ser)
In this note, we report two adult women who presented with slowly progressive retinal dysfunction in one eye with an apparent selective cone–rod pattern of loss. Though we were unable to document likely causative mutations, we find no evidence for localized alternative etiologies for the monocular visual impairments, suggesting monocular cone–rod dystrophy as a possible underlying etiology.
Case 1: A 57-year-old female presented with 4 years of progressive painless visual decline in the right eye. Her past medical history was remarkable for hypertension. On initial examination, best-corrected visual acuity was 20/50 right eye and 20/20 in the left eye. She had a relative afferent pupillary defect (rAPD) and correctly read none of the 14 Ishihara color plates with the right eye; color vision was normal in the left eye. Intraocular pressures and anterior segment examination were normal. Dilated fundus examination was significant for bilateral enlarged cup-to-disk ratios and mild macular depigmentation in the right eye.
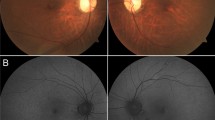
Wide-field fundus photos and fundus autofluorescence (FAF) images of both eyes were grossly unremarkable. [Fig. 2A, B]. Spectral-domain optical coherence tomography (SD-OCT) of the posterior poles showed granular loss of the outer nuclear layer and ellipsoid zone in the right eye and was within normal limits in the left eye [Fig. 2C]. SD-OCT of the retinal nerve fiber layer (RNFL) is not shown but was normal. Humphrey 24–2 visual fields showed an irregular central scotoma of the right eye and a normal field of the left eye [Fig. 2D].

Fundus A and autofluorescence B of both eyes appear within normal limits. SD-OCT of the macula C exhibits attenuation of the outer segment ellipsoid zone in the right eye (red arrow) with a reduction in the distance between the ellipsoid line and the retinal pigment epithelium underneath the fovea with an essentially normal ellipsoid zone in the left eye. Humphrey visual field 24–2 D demonstrates an irregular central scotoma in the right eye with an enlarged blind spot. The visual field of the left eye was within normal limits
To rule out mass lesions and causes of non-glaucomatous optic neuropathy, magnetic resonance imaging (MRI) of the brain and orbits with and without contrast was obtained and was within normal limits. Serologic studies of Lyme immunoglobulins, Treponema pallidum antibody, Quantiferon gold, rheumatoid factor, anti-proteinase 3, anti-myeloperoxidase, anti-aquaporin 4, anti-cyclic citrullinated protein, and angiotensin-converting enzyme were negative or within normal limits. There were no remarkable family history, history of trauma, or evidence of inflammation in either eye.
Multi-focal electroretinograms (ERGs) were obtained on the Diagnosys Espion-3 with Dawson–Trick–Litzkow (DTL) fiber electrodes (Diagnosys LLC USA, Lowell, MA). The response array had low-normal amplitude in the left eye, but was reduced by about 75% in the right eye [Fig. 3]. Upon close inspection, the response waveforms were most severely attenuated centrally. Full-field ERGs were obtained with the Diagnosys Espion-3 also using DTL fiber electrodes. Under photopic conditions, a-wave and b-wave amplitudes were greatly attenuated in the right eye, whereas responses from the left eye were within normal limits [Fig. 4]. Photopic b-wave implicit times were prolonged by 7 ms in the right eye compared with the left eye. Under scotopic conditions, the asymmetry in the response amplitudes between the two eyes was less dramatic than under photopic conditions, confirming the loss of function affecting cones more than rods [Fig. 4].

mfERGs show an extinguished response in the right eye (left column) and a low-normal response in the left eye (right column). Gray bars show normal range of ring-average amplitudes in our laboratory. Insets show response waveform arrays: In the right eye, there is a suggestion of greatest attenuation centrally

Full-field ERGs and responses to ISCEV-standard stimula (light-adapted 3.0, light-adapted 30-Hz flicker, dark-adapted 0.01, dark-adapted 3.0, dark-adapted 10.0, and dark-adapted oscillatory potentials) are shown. In each frame, responses of the right eye are shown on the left side, and responses of the left eye are shown on the right side. Each frame shows waveforms of two response events, and the averaged response of six events. Note response asymmetry, with greater relative attenuation of responses from the right eye under photopic conditions than under scotopic conditions. The latency of the response to photopic flicker is delayed as seen by comparison with the stimulus markers
Utilizing the patient’s saliva as a sample, genomic DNA was enriched for targeted regions using a hybridization-based protocol and sequenced using Illumina sequencing technology. Heterozygous variants of undetermined significance were identified in PRPH2, RCBTB1, and USH2A. Of note, the PRPH2 gene is associated with both autosomal dominant and autosomal recessive disorders including retinitis pigmentosa (MedGen UID: 334,168), Leber congenital amaurosis (MedGen UID: 137,922, macular dystrophy (MedGen UID: 1,636,950), and central areolar choroidal dystrophy (MedGen UID: 442,696).
Case 2: A 53-year-old female presented with 4 years of progressive painless visual decline in the right eye. On initial exam, best-corrected visual acuity was 20/40 right eye and 20/20 left eye. She was noted to have a trace rAPD of the right eye and correctly read none of the 14 Ishihara color plates; color vision in the left eye was normal. Intraocular pressures and anterior segment examination were normal. Dilated fundus examination was unremarkable.
Wide-field fundus photos were unremarkable. FAF showed slight hyperautofluorescence within the macula and along the arcades [Fig. 5A, B]. SD-OCT of the posterior poles showed granular loss of the ELM and EZ in the right eye and was within normal limits in the left eye [Fig. 5C]. OCT-RNFL was within normal limits. HVF 24–2 visual fields showed diffuse non-specific loss of the right eye and a normal field of the left eye [Fig. 5D].

Fundus A and autofluorescence B of both eyes appear within normal limits. There is slight hyperautofluorescence within the macula and along the arcades in the right eye compared to the left eye. SD-OCT of the macula C exhibits attenuation of the outer segment ellipsoid zone in the right eye (red arrow) and an essentially normal ellipsoid zone in the left eye. Humphrey visual field 24–2 D demonstrates a central scotoma in the right eye and an enlarged blind spot. The visual field of the left eye was within normal limits
MRI brain and orbits did not reveal mass lesions or inflammatory lesions. Laboratory testing including anti-aquaporin 4, anti-myelin oligodendrocyte glycoprotein, angiotensin-converting enzyme, and lysozyme was negative or within normal limits. There were no remarkable family history, history of trauma, or evidence of inflammation in either eye.
Multi-focal ERGs were obtained on the Diagnosys Espion-3 with DTL fiber electrodes (Diagnosys LLC USA, Lowell, MA). The response array amplitudes were normal in the left eye, but were attenuated by about 80% in the right eye [Fig. 6]. Full-field ERGs under photopic conditions showed a-wave and b-wave amplitudes that were greatly attenuated in the right eye, whereas responses in the left eye were within normal limits [Fig. 7]. Photopic b-wave implicit times were prolonged by 10 ms in the right eye compared with the left eye. Moreover, under dark-adapted conditions, response amplitudes in the right eye were less diminished, confirming the loss of function affecting primarily cones compared to rods [Fig. 7].

mfERGs show an extinguished response in the right eye (left column) and a low-normal response in the left eye (right column). Insets show response waveform arrays: In the right eye, there is a suggestion of greatest attenuation centrally

Full-field ERGs and responses to ISCEV-standard stimula (light-adapted 3.0, light-adapted 30-Hz flicker, dark-adapted 0.01, dark-adapted 3.0, dark-adapted 10.0, and dark-adapted oscillatory potentials) are shown. In each frame, responses of the right eye are shown on the left side, and responses of the left eye are shown on the right side. Each frame shows waveforms of two response events, and the averaged response of six events. Note response asymmetries and delays in the responses of the right eye
Genetic testing of the patient’s saliva sample identified a pathogenic heterozygous mutation in the BBS1 gene and a pathogenic yet low penetrance heterozygous mutation in the ABCA4 gene. Of note, the BBS1 gene is associated with autosomal recessive Bardet–Biedl syndrome and non-syndromic retinitis pigmentosa, neither of which were suspected in this patient. In addition, ABCA4 is associated with autosomal recessive inherited retinal disorders such as Stargardt disease, which again did not match this patient’s presentation.
Discussion
Cone–rod dystrophies (CRDs) are a heterogeneous group of retinal disorders characterized by dysfunction or loss of cone function and to a lesser extent, that of rods [11]. CRDs manifest after late childhood or early adulthood, and the estimated prevalence is one in 40,000.12. On dilated examination, CRDs are classically characterized by pigmentary changes within the macula. Typical symptoms include decreased visual acuity, dyschromatopsia, photophobia, decreased contrast sensitivity, and central visual field defects. As the disease progresses, nyctalopia and peripheral visual field loss can occur. With the help of ERG and FAF, the diagnosis of CRD can be made readily. On ERG, the 30 Hz flicker usually has an increased implicit time. In addition, rod and cone amplitudes may be diminished with cone responses being impacted more than rods. Occasionally, one may observe an electronegative ERG—one in which the a-wave is larger in amplitude than the b-wave [11]. FAF classically demonstrates hyperautofluorescence within the macula corresponding to areas of retinal degeneration [13]. CRDs tend to be non-syndromic, though they can be a feature of diseases such as olivopontocerebellar degeneration, amelogenesis imperfecta, Pierre-Maria ataxia among others. Mutations in 24 genes have been linked with CRD [11]. ABCA4 is implicated in 30 to 60% of autosomal recessive cases; CRX and GUCY2D in autosomal dominant cases; and RPGR in X-linked cases [12].
In recent years, there is a growing body of evidence that this group of disorders can present unilaterally and/or asymmetrically. In 1994, Sieving published a case series of two patients with suspected unilateral cone–rod dystrophy with markedly diminished cone 30-Hz flicker ERG responses. Both patients presented in their early-to-mid 60 s [5]. Nomura et al. described a case of a Japanese patient of unilateral cone dysfunction with bull’s eye maculopathy [7]. A large series by Farell described 14 cases of unilateral RP and three cases of unilateral CRD. His study found that unilateral RP comprised approximately 5% of RP cases and unilateral CRD comprises approximately 2% of cases, though these may be overestimated [6]. Many case reports have described unilateral presentations of retinitis pigmentosa, though only a few with genetic associations. Unilateral presentation of inherited retinal dystrophies such as RP may be further explained by either mosaicism or somatic mutations [8].
In our two cases, the phenotypes appeared truly unilateral without evidence of asymmetry to suggest a bilateral process. Alternative diagnoses such as trauma, medication toxicity, infection, and inflammation seemed unlikely in our cases based on patient history, laboratory testing, and clinical exam. Our patients showed findings suggestive of a selective preponderance of loss of cone function compared with rod function, with a greater disparity of ERG amplitudes between the affected and unaffected eyes under photopic conditions than under scotopic conditions. This selectivity is not typically found in most of the possible alternative diagnoses. While acquired disorders can never be entirely ruled out and it is possible that the heterozygous mutations detected through genetic testing are incidental, it also remains a possibility that the heterozygous mutations have a pathologic contribution.
With the advent of genetic diagnosis in recent decades, there has emerged a reluctance to apply the term “dystrophy” to monocular cases of the sort described here in the absence of a plausible identified mutation or at least a family history. We wish to point out that this hesitancy embodies an ironic difficulty: The presence of a likely causative mutation is no more explanatory of the absence of visual impairment in the better-seeing eye than the absence of a likely mutation is a difficulty in explaining the impairment in the poorer-seeing eye, particularly in light of the limitations in our ability to identify causative mutations even in clear cases of inherited retinal disease [14]
Writing in 1963, Franceschetti and colleagues summarized their discussion of unilateral pigmentary retinopathy as follows: “There is no longer any reason to doubt the existence of unilateral pigmentary retinopathy, particularly since the introduction of electroretinography. With the aid of this test, definite diagnosis can now be made immediately, and a long period of observation is no longer necessary. It seems likely that the number of authentic unilateral cases will increase in the future” [2]. While it is unclear whether ERG, or our other enhanced diagnostic tools have eliminated our reticence regarding these unilateral cases, perhaps we should at least continue to entertain the possibility that the pathophysiology in these cases is more similar than different from that of bilateral cases with similar anatomic and physiologic findings.
References
Francois J, Verriest G (1952) Retinopathie pigmentaire unilaterale. Ophthalmologica 124:65–88. https://doi.org/10.1159/000301252
Franceschetti A, Francois J, Babel J (1974) Chorioretinal Heredodegenerations: an updated report of La Société d'Ophtalmologie. Springfield, IL: Charles C Thomas, pp. 266–274
Kolb H, Nr G (1964) Three cases of unilateral pigmentary degeneration. Br J Ophthalmol 48(9):471–479. https://doi.org/10.1136/bjo.48.9
Errea MH, Robson AG, Wong T et al (2019) Unilateral pigmentary retinopathy: a retrospective case series. Acta Ophthalmol 97:e601–e617. https://doi.org/10.1111/aos.13981
Sieving PA (1994) Unilateral cone dystrophy’: ERG changes implicate abnormal signaling by hyperpolarizing bipolar and/or horizontal cells. Trans Am Ophthalmol Soc 92:459–474
Farrell DF (2009) Unilateral retinitis pigmentosa and cone–rod dystrophy. Clin Ophthalmol 3:263–270. https://doi.org/10.2147/opth.s5130
Nomura R, Kondo M, Tanikawa A, Yamamoto N, Terasaki H, Miyake Y (2001) Unilateral cone dysfunction with bull’s eye maculopathy. Ophthalmology 108(1):49–53. https://doi.org/10.1016/s0161-6420(00)00450-4
Marsiglia M, Duncker T, Peiretti E, Brodie SE, Tsang SH (2012) Unilateral retinitis pigmentosa: a proposal of genetic pathogenic mechanisms. Eur J Ophthalmol 22(4):654–660. https://doi.org/10.5301/ejo.5000086
Sim PY, Jeganathan VSE, Wright AF, Cackett P (2018) Unilateral retinitis pigmentosa occurring in an invidiaul with a mutation in the CLRN1 gene. BMJ Case Rep. https://doi.org/10.1136/bcr-2017-222045
Mukhopadhyay R, Holder GE, Moore AT, Webster AR (2011) Unilateral retinitis Pigmentosa occurring in an individual with a germline mutation in the RP1 gene. Arch Ophthalmol 129(7):954–956. https://doi.org/10.1001/archophthalmol.2011.171
Puech B, De Laey JJ, Holder GE (2014) Inherited Chorioretinal dystrophies: a textbook and atlas. Springer, Heidelberg
Hamel CP (2007) Cone rod dystrophies. Orphanet J Rare Dis 2:7. https://doi.org/10.1186/1750-1172-2-7
Lima LH, Zett C, Kniggendorf V et al (2018) Progressive expansion of the hyperautofluorescent ring in cone–rod dystrophy patients. Ophthalmic Genet 39(4):492–499. https://doi.org/10.1080/13816810.2018.1461911
Stone EM, Andorf JL, Whitmore SS (2017) Clinically focused molecular investigation of 1000 consecutive families with inherited Retinal disease. Ophthalmology 124(9):1314–1331. https://doi.org/10.1016/j.ophtha.2017.04.008
Funding
The authors declared that they have no funding.
Author information
Authors and Affiliations
Contributions
All authors attest that they meet the current ICMJE criteria for authorship.
Corresponding author
Ethics declarations
Conflicts of interest
The following authors have no financial disclosures: SP, AN, VG, SG, SB.
Informed consent
Informed consent was obtained from the participants included in this study. The participants have consented to the submission of the case report to the journal.
Statement of human rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of NYU Langone Health and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Statement on the welfare of animals
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Choi, S., Pandit, S.A., Nair, A.A. et al. Two cases of unilateral cone–rod dysfunction presenting in adult females. Doc Ophthalmol 145, 271–281 (2022). https://doi.org/10.1007/s10633-022-09893-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10633-022-09893-9