
Phytochemical investigation of the stem bark of Xeromphis nilotica led to the isolation of two new triterpenoid saponins; 3-O-β-D-glucopyranosyl (1→3)-β-D-glucopyranosyl-olean-12-ene-23,27-diol (1) and 3-O-{β-D-glucopyranosyl-(1→3)-O-[β-D-glucopyranosyl-(1→3)]-β-D-glucopyranosyl}-oleanolic acid (2). Their structures were elucidated through extensive chromatographic separations and spectroscopic methods [1D, 2D NMR and HR-ESI-MS]. Sugar moieties were identified through TLC after acid hydrolysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Xeromphis nilotica (Stapf) Keay (local name, Shagarat Almarfaein) is a Sudanese medicinal plant, commonly used in western Darfur and the Nuba Mountains area for treatment of jaundice and as a fish poison [1, 2]. It is also reputed to have a medicinal value for the treatment of various diseases including epilepsy, pain, mental disorder and fever [3, 4]. Pharmacological investigation of X. nilotica material reveals important biological activities such as antinociceptive, anti-inflammatory [5], molluscicidal [6], antischistosomal [7], and CNS activities [8, 9]. Previous phytochemical investigation of X. nilotica roots, stem barks and leaves resulted in the isolation of a number of aromatic compounds, particularly simple coumarin derivatives, iridoid [2], flavonoids, carbohydrates, alkaloids and triterpenoid saponins [1, 6, 10].
Our previous investigations of this species led to the isolation and characterization of coumarin glycosides [11] and triterpene saponins [12]. The following contribution is based on our continuing investigations on the chemical constituents of the stem barks of X. nilotica. In this paper, we report the structural determination of two new oleanolic saponins; 3-O-β-D-glucopyranosyl-(1→3)-β-D-glucopyranosylolean-12-ene-23,27-diol (1) and 3-O-{β-D-glucopyranosyl-(1→3)-O-[β-D-glucopyranosyl-(1→3)]-β-D-glucopyranosyl}-oleanolic acid (2).
Compound 1 was obtained as a white amorphous powder. The molecular formula of 1 was found to be C42H70O13NH4 via the positive HR-ESI-MS ion at m/z 800.4463 [M + NH4]+. The 1H NMR experiment data of 1 (Table 1), revealed six tertiary methyl protons at δ 0.83 (3H, s, H-24), 1.04 (3H, s, H-25), 1.14 (3H, s, H-26), 0.80 (3H, s, H-28), 0.89 (3H, s, H-29), and 0.94 (3H, s, H-30); one olefinic proton at δ 5.22 (1H, br.s, H-12); two anomeric protons at δ 4.36 (1H, d, J = 7.8 Hz) and 4.55 (1H, d, J = 7.8 Hz) together with one signal at δ 2.84–2.86 (1H, m, H-18) and one signal at δ 3.18 (1H, dd, J = 11.7, 4.4 Hz, H-3). 13C NMR spectrum data of 1 (Table 2) showed 42 carbon resonances, 30 of the 42 carbons were assigned to the triterpenoid skeleton and 12 to the two sugars moiety. They included six tertiary carbons at δ 14.5 (C-24), 15.5 (C-25), 16.3 (C-26), 32.1 (C-28), 33.3(C-29), and 22.5 (C-30); two olefinic carbons at δ 122.1 (C-12) and 143.4 (C-13), and two anomeric protons at δ 104.4 (C-1′) and 103.7 (C-1′′), together with three oxygenated methine carbons at δ 62.6 (C-27), 63.6 (C-23), and 89.4 (C-3).
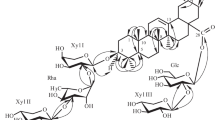
Overall 1H and 13C NMR data of 1 (Tables 1 and 2) showed great similarity to olean-2-en-27-ol [13], except that C-23 and an C-3 were attached with a hydroxyl group and sugar chain, respectively, in 1. Acid hydrolysis of 1 afforded sugar moiety which was identified by TLC as glucose. The HMBC spectrum (Fig. 1), showed the correlation between H-1′′ (δ 4.55) of the terminal glucose with C-3′ (δ 86.4) of the inner glucose and H-1′ (δ 4.36) of the inner glucose with C-3 (δ 89.4) of aglycone.

The key HMBC and 1H–1H COSY correlations observed in compounds 1 and 2.
The configuration of the sugars was established to be β glucose units characterized by the large coupling constants [14, 15], starting from the anomeric proton at δ 4.36 (d, J = 7.8 Hz) and the other anomeric proton resonating at δ 4.55 (d, J = 7.8 Hz). On the basis of all the foregoing information, compound 1 was identified as 3-O-β-D-glucopyranosyl (1→3)-β-Dglucopyranosylolean-12 ene-23,27-diol.
Compound 2 was obtained as a white amorphous powder. HR-ESI-MS, showed peak at m/z 965 [M + Na]+, which corresponds to the molecular formula C48H78O18. Detailed analysis of the 1H NMR spectrum data of 2 (Table 1) exhibited seven methyl protons singlets at δ 1.13 (3H, s, H-23), 0.82 (3H, s, H-24), 0.93 (3H, s, H-25), 1.04 (3H, s, H-26), and 1.88 (3H, s, H-27), 1.23 (3H, s, H-29), 1.24 (3H, s, H-30); an olefinic proton as a broad singlet at δ 5.19 (1H, br.s, H-12); three anomeric protons δ 4.34 (d, J = 7.8 Hz, H-1′); 4.57 (d, J = 7.8 Hz, H-1′′) and 5.16 (d, J = 7.6 Hz, H-1′′′) together with a multiplet of one proton at δ 2.87–2.89 (1H, m, H-18) and one signal at δ 3.17 (1H, dd, J = 11.6, 4.5 Hz, H-3).
The 13C NMR spectrum data (Table 2) showed seven methyl carbons signals at δ 29.3 (C-23), 15.7 (C-24), 14.6 (C-25), 16.8 (C-26), 26.2 (C-27), 32.8 (C-29), and 23.5 (C-30); two olefinic carbon signals at δ 121.2 (C-12) and 145.1 (C-13); downfield signal at δ 183.8 (C-28), which presented carboxylic carbon and three anomeric carbons resonating at δ 104.9 (C-1′), 103.7 (C-1′′), and 101.2 (C-1′′′) attributed to three sugar residues. Acid hydrolysis suggested that the monosaccharide of this compound is D-glucose, which was identified by TLC analysis of its Rf value [16]. The aglycone was further recognized to be oleanolic acid by comparison of its 1H and 13C NMR data with those reported in the literature [14] which were in good agreement. The β-configuration of the anomeric position of the glucosyl units was deduced from the large values of the coupling constant of the H-1′, H-1′′, and H-1′′′ (J = 7.8 Hz; J = 7.8 Hz, and J = 7.6 Hz), respectively, the downfield shifts of C-3′ (86.5) and C-3′′ (82.6) of the first and second glucosyl moieties suggested the bonding points of the trisaccharide chain [17, 18]. The key HMBC spectrum data (Fig. 1), showed correlations between the anomeric proton at δH 5.16 (H-1′′′) of the terminal glucose with inner glucose at δC 82.6 (C-3′′); the anomeric proton of the inner glucose at δH 4.57 (H-1′′) with the first glucose at δC 86.5 (C-3′) and anomeric proton of the first glucose at δ 4.34 (H-1′) with the aglycone part at δC 89.6 (C-3). On the basis of this evidence, the structure of compound 2 elucidated as 3-O-{β-D-glucopyranosyl-(1→3)-O-[β-D-glucopyranosyl-(1→3)]-β-D-glucopyranosyl}-oleanolic acid.
Experimental
General Experimental Procedures. Melting points were recorded on an X-4 type micro-melting point apparatus, which was uncorrected. The NMR spectra were measured on a Bruker-DRX-400-NMR spectrometry and with TMS as internal standard, HR-ESI-MS were obtained with API QSTAR Pulsari System Mass Spectrometer. Column chromatography was carried out on silica gel (Merck Kiesel gel 300–400 mesh), TLC was performed with silica gel GF254 (Merck). All the chemicals and solvents were commercial grade and used after further purification.
Plant Material. “Shagarat Almarfaein” stem barks were collected from the Zalingei area, central Darfur State, Sudan, in April of 2017, and identified by Prof. G. A. Yagoub, Faculty of Agriculture, University of Zalingei. Voucher specimens (No. 20171013), was deposited in the herbarium of the author′s laboratory.
Extraction and Isolation. The dried stem bark of Xermophis nilotica (1.5 kg) was powdered and extracted with 95% EtOH at room temperature (every 7 days × 3 L). After evaporation of the solvent under vacuum (Rotary evaporation), the dried residue (355 g) was suspended in H2O and then partitioned with EtOAc. The EtOAc fraction (120 g) was chromatographed over silica gel (Merck Kiesel gel 300–400 mesh) using (CHCl3–MeOH) gradually, first eluted with pure CHCl3, then checked by thin layer chromatography (TLC) using the CHCl3–MeOH (10:1, 10:2, 10:3) then with CHCl3–MeOH (10:1–1:1) and finally eluted with pure MeOH (33 fractions were collected) as mobile phases. Fractions showing similarities on TLC were combined together to provide three fractions (FI, FII, and FIII). Fraction FIII (35 g) was loaded on a silica gel column eluting with EtOAc, increasing polarity by adding MeOH, to yield two subfractions (Fi, Fii). Subfraction Fi was further subjected to column chromatography (CC) again using EtOAc and MeOH mixtures with increasing polarities to give compound 1, while subfraction Fii was re-chromatographed over silica to obtain compound 2.
3-O-β-D-Glucopyranosyl (1→3)-β-D-glucopyranosylolean-12 ene-23,27-diol (1), white amorphous powder. UV 365 nm showed blue color, deep purple color after spraying 7% H2SO4 reagent, molecular formula C42H70O13NH4. HR-ESI-MS m/z 800.4463 [M + NH4]+. 1H (400 MHz, MeOH-d4) and 13C (100 MHz, MeOH-d4) NMR spectral data, see Tables 1 and 2.
3-O-{β-D-Glucopyranosyl-(1→3)-O-[β-D-glucopyranosyl-(1→3)]-β-D-glucopyranosyl}-oleanolic acid (2), white amorphous powder. UV 365 nm showed blue color and deep purple color after spraying 7% H2SO4 reagent. HR-ESI-MS, showed peaks at m/z 965.5073 [M + Na]+, corresponding to formula C48H78O18Na. 1H (400 MHz, MeOH-d4) and 13C (100 MHz, MeOH-d4) NMR spectral data, see Tables 1 and 2.
Acid Hydrolysis of the Saponins. 10 mg each of compound 1 and 2 was added to a solution of 10% AcOH–EtOH (10 mL). The mixture was stirred for 6 h. After cooling, the solution was removed under reduced pressure. The residue was diluted with H2O (5 mL), and the resulting precipitate was collected and chromatographed on a silica gel column (35 g, Si gel). Elution of the column with 10% MeOH in CHCl3 afforded the aglycones. The aqueous phase was neutralized by NaHCO3 and analyzed for sugars using PC. The solvent system used was n-BuOH–AcOH–H2O, 4:1:5 [15].
References
A. K. Bashir, J. Pharmacogn. Phytochem., 34, 202 (1996).
H. A. R. Farid, O. Kuner, E. Haslinger, and C. Seger, Monatshefte fur Chemie, 133, 1453 (2002).
H. Inga, O. Hedberg, P. J. Madati, K. E. Mshigeni, E. N. Mshiu, and G. Samuelsson, J. Ethnopharmacol., 9, 237 (1983).
S. Margaret, Flowering Plants in West Africa, Cambridge University Press, 1988.
B. Adzu, M. B. Amizan, and S. E. Okhale, J. Ethnopharmacol., 158, 271 (2014).
Y. E. El Kheir and M. H. Salih, Fitoterapia, 51, 143 (1980).
S. M. Sulaiman, A. K. Bashir, and A. M. Mohamed, Int. J. Crude Drug Res., 26, 17 (1988).
B. Adzu, M. B. Amizan, A. A. Njan, J. O. C. Ezeowumelu, and D. D. Akumka, IJBCS, 2, 359 (2008).
N. M. Danjuma, A. U. Zezi, A. H. Yaro, A. M. Musa, A. Ahmed, H. A. Sanni, and I. M. Maje, Int. J. Appl. Res. Nat. Prod., 2, 5 (2009).
E. Lemmich, C. Cornett, P. Furu, C. L. Jorstian, A. D. Knudsen, C. Olesen, A. Salih, and S. T. Thiilborg, Phytochemistry, 39, 63 (1995).
I. Abdurrahman, Y. C. Xia, and H. Y. Lai, WJPR, 15, 292 (2016).
I. A. Adam, I. Omer, and H. Yu-Lai, J. Med. Chem. Sci., 1, 41 (2018).
B. S. Siddiqui, F. Ilyas, M. Rasheed, and S. Begum, Phytochemistry, 65, 2079 (2004).
A. A. Magid, L. V. Nazabadioko, I. Renimel, D. Harakat, C. Moretti, and C. Lavaud, Phytochemistry, 67, 2098 (2006).
M. A. Ouyang, Y. Q. Liu, H. Q. Wang, and C. R. Yang, Phytochemistry, 49, 2485 (1998).
A. J. Harborne, Phytochemical Methods a Guide to Modern Techniques of Plant Analysis, Springer Science & Business Media, 1998.
L. Voutquenne, P. Guinot, O. Thoison, T. Sevenet, and C. Lavaud, Phytochemistry, 64, 781 (2003).
J. Li, X. Huang, X. H. Jiang, Q. F. Zhu, Y. Yang, and G. C. Gao, Chem. Pharm. Bull., 63, 389 (2015).
Acknowledgment
The authors are grateful to the China Scholarship Council (CSC) for the research scholarship, and to the Department of Botany, Faculty of Agriculture, University of Zalingei for facilitating the collection and authentication of the plant material.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 2, March–April, 2023, pp. 270–273.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Adam, I.A., Cai-Xia, Y. Two New Oleanolic Saponins from Stem Barks of Xeromphis nilotica. Chem Nat Compd 59, 318–322 (2023). https://doi.org/10.1007/s10600-023-03984-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-023-03984-y