
For the first time, the n-hexane extract from the dried stem bark of Cassia grandis L.f. was investigated, and one new fatty acid derivative, named casgraester A (1), was obtained, together with three known ones, 2,3-dihydroxypropyl 28-hydroxyoctacosanoate (2), 2,3-dihydroxypropyl octacosanoate (3), bis-(2,3-dihydroxypropyl)tetracosanedioate (4), three known triterpenoids, lupeol (5), betulinic acid (6), oleanolic acid (7), and three known steroids, β-sitosterol (8), 3-O-β-D-glucopyranosyl-β-sitosterol (9), and 3-O-β-D-glucopyranosylbrassicasterol (10). For the first time, compounds 2–4, 6, and 10 were reported from the genus Cassia, while compounds 5, 7, and 9 were found for the first time from this species. Their structures were elucidated by IR, HR-ICR-MS, ESI-MS/MS, and NMR experiments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cassia grandis belongs to the Caesalpiniaceae family. In Vietnam, it is a medicinal plant and has been used for the treatment of skin disorders, back pain, and aches [1]. It is used to treat anemia in Costa Rica and as an expectorant or astringent in Guetemala [2]. Continuing a study of the bioactive compounds from medicinal plants in Vietnam [3,4,5,6,7,8] and from the leaves of this species [9], in this paper we study the isolation and structural elucidation of one new fatty acid derivative, named casgraester A (2,3-dihydroxypropyl 25-hydroxypentacosanoate) (1), three known fatty acid derivatives, 2,3-dihydroxypropyl 28-hydroxyoctacosanoate (2) [4], 2,3-dihydroxypropyl octacosanoate (3) [10], and bis-(2,3-dihydroxypropyl)tetracosanedioate (4) [11]; three known triterpenoids, lupeol (5) [12], betulinic acid (6) [13], and oleanolic acid (7) [14], and three known steroids, β-sitosterol (8) [15], 3-O-β-D-glucopyranosyl-β-sitosterol (9) [16], and 3-O-β-D-glucopyranosylbrassicasterol (10) [17] from the n-hexane extract of Cassia grandis stem bark collected in Can Tho city, Vietnam. For the first time, the n-hexane extract from the dried stem bark of Cassia gandis L.f. was investigated. Compounds 2–4, 6, and 10 were reported for the first time from the genus Cassia, while compounds 5, 7, and 9 were found for the first time from this species.

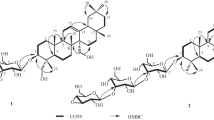
Compound 1 was obtained as a white amorphous powder. The molecular formula was established as C28H56O5 from HR-ICR-MS data ([M + H]+m/z 473.42061, calcd for 473.42060). The IR spectrum of 1 showed absorptions of hydroxyl (3422 cm–1) and carbonyl (1735 cm–1) groups. The 13C NMR and DEPT spectrum of 1 showed 28 carbons, including one carbonyl carbon, one oxygenated methine carbon, three oxygenated methylene carbons, and 23 methylene carbons. The presence of one oxymethine carbon at δ 70.0 (C-2′) and two oxymethylene carbons at δ 65.1 (C-1′) and 63.2 (C-3′) corresponded to one oxymethine proton at δ 3.86–3.89 (1H, m, H-2′) and four oxymethylene protons at δ 3.63–3.66 (2H, m, H-3′) and 4.09–4.14 (2H, m, H-1′), respectively, indicating an sn-1-monoacylglycerol framework [17]. In addition, it also revealed one carbonyl carbon at δ 174.5 (C-1), one α-methylene carbon at δ 34.1 (C-2), one β-methylene carbon at δ 24.8 (C-3), one ω3-methylene carbon at δ 25.7 (C-23), one ω2-methylene carbon at δ 32.5 (C-24), and one oxymethylene carbon at δ 62.6 (C-25), characteristic of an ω-hydroxy fatty acid [18]. The HMBC spectrum showed a correlation between two oxymethylene protons at δ 4.09–4.14 (2H, m, H-1′) and a carbonyl carbon at δ 174.5 (C-1), so the fatty acid chain is linked to the glycerol framework at C-1′. On the other hands, the HR-ICR-MS data of 1 showed that the long-chain fatty acid was 25-hydroxypentacosanoic acid. Further, an ω-hydroxy fatty acid was also confirmed by an ion peak at m/z 397.30 [C25H50O3 – H]– in the ESI-MS/MS spectrum. Based on data of HR-ICR-MS, ESI-MS/MS, and 1D and 2D NMR and comparison with previous published data [18], the structure of 1 was identified as 2,3-dihydroxypropyl 25-hydroxypentacosanoate, named casgraester A.
Experimental
General. The melting point was recorded on an Electrothermal 9100 UK melting point meter. The optical rotation was measured on an ADP220 polarimeter (Bellingham + Stanley Ltd., RG224BA, UK). The IR data were recorded on an FT-IR spectrometer (NICOLET-6700, USA). The high-resolution ion cyclotron resonance mass spectrometric analysis (HR-ICR-MS) was performed on a high-performance liquid-chromatography-mass spectrometer system (Varian 910-MS TQFTMS, Germany). The electrospray ionization mass spectrometric analysis (ESI-MS/MS) was performed on a liquid-chromatographymass spectrometer system (HP 1100 series, LC/MSD trap, Agilent, USA). The 1H NMR (500 MHz), 13C NMR (125 MHz), DEPT, HSQC, and HMBC spectra were recorded on a Bruker AM500 FT-NMR spectrometer using tetramethylsilane (TMS) as internal standard. Normal and reversed phase chromatography were carried out using silica gel 230–400 mesh (Merck KGaA, 64271 Darmstadt, Germany). Analytical TLC was carried out on silica gel plates (Kieselgel 60 F254, Merck). Compounds were visualized by spraying with aqueous 10% H2SO4 and heating for 3–5 min.
Plant Material. The stem bark of C. grandis was collected in Can Tho City, Vietnam in March 2012; and identified by Dr. Minh Quan Dang, Can Tho University. A voucher specimen (No. VH/LUAN-0402) was deposited in the Institute of Chemical Technology, Vietnam Academy of Science and Technology.
Extraction and Isolation. The dry powdered stem bark of Cassia grandis L.f. (17 kg) was extracted with 96% EtOH three times (3 × 10 L, total amount 30 L) at room temperature. The residue was filtered, the solvents were removed under low pressure, and a crude extract (795 g) was obtained. To the crude extract was added distilled water and the whole subjected to liquid-liquid extraction and successively partitioned into the n-hexane fraction (230 g), the EtOAc (145 g) fraction, and the aqueous fraction (410 g). The EtOAc subfraction was subjected to silica gel column chromatography (CC). The mobile phase was n-hexane–EtOAc, 95:5, 90:10, 85:15, 80:20, 75:25, 70:30, 50:50, 25:75, 0:100. Nine fractions were obtained (Frs. BE1–BE9). Fractions BE2 (13.6 g) and BE3 (22.8 g) were again subjected to CC twice with n-hexane–EtOAc solvent systems as eluent in the direction of increasing polarization. Finally, 130 mg of compound 8 were obtained from Fr. BE2, and 650 mg of compound 6 were obtained from Fr. BE3. Fraction BE4 (26.4 g) was also subjected to CC two or three times with chloroform–methanol solvent systems as eluent in the direction of increasing polarization. Finally, 660 mg compounds 7, 1.2 g 9, and 440 mg 10 were obtained. Fraction BE5 (24.7 g) was again subjected to CC several times with chloroform–methanol solvent systems as eluent to yield 220 mg compounds 1, 20 mg 2, 18 mg 3, 10 mg 4, and 26 mg 5.
Casgraester A (1). White amorphous powder, mp 132–135°C, \( {\left[\upalpha \right]}_{\mathrm{D}}^{25} \) +10.5° (0.01 M, CHCl3). IR (KBr, νmax, cm–1): 3422, 2848, 1735, 1467, 1178. HR-FT-ICR-MS m/z 473.42061 [M + H]+. ESI-MS/MS m/z 471.30, 397.30. 1H NMR (500 MHz, CDCl3 and CD3OD, δ, ppm): 1.26–1.29 (40H, br.s, H-4→23), 1.54–1.57 (2H, m, H-24), 1.58–1.65 (2H, m, H-3), 2.33–2.36 (2H, m, H-2), 3.54–3.61 (2H, m, H-25), 3.63–3.66 (2H, m, H-3′), 3.86–3.89 (1H, m, H-2′), 4.09–4.14 (2H, m, H-1′). 13C NMR (125 MHz, CDCl3 and CD3OD, δ, ppm): 24.8 (C-3), 25.7 (C-23), 29.1–29.6 (C-4→22), 32.5 (C-24), 34.1 (C-2), 62.6 (C-25), 63.2 (C-3′), 65.1 (C-1′), 70.0 (C-2′), 174.5 (C-1).
References
T. L. Do, Medicinal Plants and Remedy of Vietnam, Publisher of Medicine, Hanoi, Vietnam, 2004.
E. Valencia, A. Madinaveitia, J. Bermejo, and M. P. Gupta, Fitoterapia, 66, 476 (1995).
T. N. Ngo, C. T. Vo, N. K. T. Pham, N. M. Phan, T. D. Bui, Q. L. Ngo, V. S. Dang, C. L. Tran, D. T. Mai, and T.P. Nguyen, Nat. Prod. Res., 33, 174 (2019).
T. P. Nguyen, T. D. Le, N. M. Phan, T. D. Bui, N. K. T. Pham, T. M. L. Do, D. T. Nguyen, and D. T. Mai, Nat. Prod. Res., 30, 2389 (2016).
T. P. Nguyen, D. T. Mai, T. H. T. Do, and N. M. Phan, Nat. Prod. Commun., 12, 1061 (2017).
T. P. Nguyen, N. M. Phan, T. D. Bui, T. D. Le, N. M. A. Tran, T. N. T. Pham, and D. T. Mai, Nat. Prod. Res., 31, 2281 (2017).
T. P. Nguyen, C. L. Tran, C. H. Vuong, T. H. T. Do, T. D. Le, D. T. Mai, and N. M. Phan, Nat. Prod. Res., 31, 2587 (2017).
T. P. Nguyen, T. T. V. Tran, D. T. Mai, T. D. Le, N. M. Phan, and T. D. Bui, Nat. Prod. Res., 29, 1432 (2015).
P. T. N. Trinh, N. Q. Luan, M. D. Tri, V. D. Khanh, N. H. An, P. N. Minh, P. N. An, N. T. L. Thuy, N. K. P. Phung, and L. T. Dung, Nat. Prod. Res., 31, 1733 (2017).
C. D. R. Jose, P. Pipijn, and G. Ana, J. Cereal Sci., 58, 248 (2013).
N. Y. Yang, W. W. Tao, and J. A. Duan, J. Chem. Res., 7, 423 (2009).
A. K. Jamal, W. A. Yaacob, and L. B. Din, J. Phys. Sci., 19, 45 (2008).
D. Ghias, B. S. S. Waliullah, A. Muhammad, S. Anwar, A. Ashfaq, and U. Ala, Middle-East J. Sci. Res., 8, 85 (2011).
W. A. A. Yahya, W. A. Yaacob, and I. Nazlina, Malaysian J. Anal. Sci., 15, 22 (2011).
N. L. H. Ton, M. H. Nguyen, and D. L. Tran, Cantho Univ. J. Sci., 19b, 56 (2011) (in Vietnamese).
J. Gao, L. Hu, and J. Liu, Steroids, 66, 771 (2001).
R. Sacchi, F. Addeo, and L. Paolillo, Magn. Reson. Chem., 35, S133 (1997).
R. K. Mukherjee, Y. Fujimoto, and K. Kakinuma, Phytochemistry, 37, 1641 (1994).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 3, May–June, 2020, pp. 341–342.
Rights and permissions
About this article

Cite this article
Ngo, Q.L., Phan, N.M. & Nguyen, T.P. New Fatty Acid Derivative from the Stem Bark of Cassia grandis. Chem Nat Compd 56, 392–394 (2020). https://doi.org/10.1007/s10600-020-03044-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-020-03044-9