

Pyrazolo[1,5-a]pyrazin-4(5H)-ones react regioselectively with N-iodosuccinimide in the presence of N-methylmorpholine to form 7-iodo derivatives. Their carbonylation catalyzed with Pd(dppf)Cl2 under pressure in MeOH solution yielded methyl 4-oxo-4,5-dihydropyrazolo-[1,5-a]pyrazine-7-carboxylates, which were transformed into the corresponding carboxylic acids by alkaline hydrolysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Pyrazolo[1,5-a]pyrazin-4(5H)-ones are important derivatives of a corresponding biheteroaromatic system due to their role as basic substrates for various synthetic transformations aimed at creating biologically active compounds.1 These compounds have been extensively studied in the biomedical field, leading to the discovery of vasopressin1b,j,k and fibrinogen2,3 receptor antagonists, selective positive allosteric glutamate receptors GluN2A4 and mGluR55 modulators, inhibitors of mycobacterium tuberculosis H37RV,6 inhibitors of lung cancer tumors A549 and H322b,7,8 inhibitors of catalytic activity of HIV-19 integrase, TANKs and polyADP-ribose polymerase PARP-1.10 Recently, certain derivatives of pyrazolo[1,5-a]pyrazin-4(5H)-one have been proposed for the treatment of lysosomal and neurodegenerative,11 as well as cardiovascular12 diseases.
The analysis of bioactive compounds in the pyrazolopyrazinone series has revealed that most of them contain functional substituents at positions 2 and 5, which seem to play a significant role in their pharmacological profile. However, it is worth mentioning that derivatives of this heterocyclic system with functional groups at positions 6 and 7 have received limited attention so far, thereby restricting its medical and biological potential. Consequently, we aimed to develop a convenient method to obtain previously unexplored pyrazolo[1,5-a]pyrazin-4(5H)-ones with synthetically potent methoxycarbonyl and carboxyl groups at position 7. We believe that incorporating these groups into the pharmacophore of pyrazolopyrazinone scaffold represents the most efficient approach for designing new synthetic platforms with potential significance in combinatorial and medicinal chemistry.
The proposed solution to address this issue involved a sequential structural modification of the pyrazolopyrazinone fragment, with the key step being the selective iodination of position 7 in pyrazolo[1,5-a]pyrazinones 1a–d. This task is rather challenging due to the combination of pyrazine and pyrazole nuclei in these structures, with the latter exhibiting a more pronounced nucleophilic center. Patent data also indicate that the electrophilic halogenation reaction of 4-substituted pyrazolo[1,5-a]pyrazines using N-halogensuccinimides primarily occurs at position 3 of the bicyclic system.1c,h,13 Additionally, the iodination of nonactivated pyrazine rings in their azoloannulated analogs, such as imidazo[1,5-a]pyrazines,14 oxazolo[4,5-b]pyrazines,15 and triazolo[4,3-e]pyrazines,16 typically requires prior metallation and lacks high selectivity.
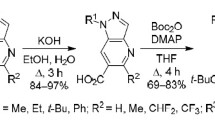
In screening experiments conducted using pyrazolopyrazinone 1a as a representative example, it was observed that the optimal reaction with N-iodosuccinimide (NIS) occured at room temperature in a DMF solution in the presence of N-methylmorpholine as a base. These conditions facilitate a regioselective process, leading to the formation of 7-iodo derivative 2a in 86% yield. Under the similar conditions, iodination of compounds 1b–d led to derivatives 2b–d in 79–92% yields (Scheme 1).

Scheme 1
The structure of the synthesized 7-iodo-substituted pyrazolo[1,5-a]pyrazin-4-ones 2a–d was confirmed by 1H NMR using the steady-state NOE technique for NH and H-6 proton irradiation. Compound 2b was utilized as an example for this analysis.
In organic chemistry, the ester group is a well-known functional substituent with a wide range of synthetic applications. Among the various methods for introducing ester groups into aromatic and heterocyclic compounds, Pd-catalyzed carbonylation reactions of aryl halides and related compounds hold a significant place.17 Interestingly, such transformations have not been previously investigated in iodinated pyrazines and their condensed derivatives, although there is data on similar reactions involving structurally related 4-chloropyrazolo[1,5-a]pyrazines.18
Our research revealed that 7-iodopyrazolo[1,5-a]pyrazin-4-ones 2a–d can undergo carbonylation in a MeOH solution in an autoclave at elevated pressure (20 atm) and temperature (100°C) in the presence of catalytic amounts of Pd(dppf)Cl2 and more than double excess of Et3N. Exposure of the reaction mixture for 16 h was sufficient to obtain methyl carboxylates 3a–d in 67–84% yield (Scheme 1). Conversion of the latter to acids 4a–d completed after 8 h boiling in an aqueous KOH solution. Despite the relatively harsh reaction conditions, undesired decarboxylation did not take place so that the target products formed in 90–95% yields.
In summary, novel 7-iodopyrazolo[1,5-a]pyrazin-4-ones were successfully synthesized. Their utility as convenient reagents for the palladium-catalyzed synthesis of methyl 4-oxopyrazolo[1,5-a]pyrazine-7-carboxylates was demonstrated. These compounds, along with their corresponding acids, serve as vital building blocks for the construction of potentially bioactive compounds. The development of these scaffolds provides valuable opportunities for further exploration and design of molecules with potential pharmacological applications.
Experimental
IR spectra were recorded on a Bruker Vertex 70 in KBr pellets. 1H and 13C NMR spectra were recorded on Varian VXR-400 (400 and 126 MHz, respectively, compounds 1c, 2a–d, 3, 4 a,b,d) and Varian Mercury 300 (300 and 76 MHz, respectively, compounds 3, 4 c) spectrometers in DMSO-d6, internal standard TMS. The NOE experiment was performed on an Agilent ProPulse 600 spectrometer (600 MHz) in DMSO-d6. Mass spectra were recorded on an Agilent LC/MSD SL instrument (Zorbax SB-C18 column, 4.6 × 15 mm, 1.8 μm (PN 82(c)75-932)), solvent DMSO, electrospray ionization. Elemental analysis was performed on a PerkinElmer CHN-analyzer 2400 series instrument in the Analytical laboratory of the Institute of Organic Chemistry of the National Academy of Sciences of Ukraine. The melting points were determined on a Kofler table and were not corrected.
All operations with CO were performed in a well-ventilated fume hood with an Orvibo Zigbee 3V (SP20-0) carbon monoxide detector. The Pd(dppf)Cl2 catalyst was provided by Enamine Ltd. (Kyiv, Ukraine). The starting compounds 1a,b1d and 1d1e were obtained according to the literature methods.
2-Ethylpyrazolo[1,5-a]pyrazin-4(5H)-one (1c) was prepared from 2-ethyl-7-hydroxy-6,7-dihydropyrazolo[1,5-a]-pyrazin-4-(5H)-one (5.4 g, 30 mmol, provided by Enamine Ltd.) following the literature method.1d Yield 4.2 g (86%), yellow powder, mp 207–208°C. IR spectrum, ν, cm–1: 1676 (C=O), 3331 (NH). 1H NMR spectrum, δ, ppm (J, Hz): 1.24 (3H, t, J = 7.8, CH3); 2.70 (2H, q, J = 7.8, CH2); 6.76 (1H, d, J = 6.0, H-6); 6.79 (1H, s, H-3); 7.55 (1H, d, J = 6.0, H-7); 11.09 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 14.1; 21.3; 102.6; 110.4; 115.7; 134.5; 155.7; 155.8. Mass spectrum, m/z (Irel, %): 164 [M+H]+ (100). Found, %: C 58.78; N 5.36; N 25.75. C8H9N3O. Calculated, %: C 58.88; H 5.56; N 25.75.
Synthesis of compounds 2a–d (General method). NIS (31.5 g, 0.14 mol) was added portionwise to a solution of pyrazolopyrazinone 1a–d (0.1 mol) and N-methylmorpholine (10.1 g, 0.1 mol) in DMF (150 ml) at room temperature. The reaction mixture was stirred for 12 h, the solvent was evaporated, H2O (250 ml) was added to the solid residue, and the resulting precipitate was filtered off, washed with H2O (100 ml), MeCN (50 ml), and dried in air.
7-Iodopyrazolo[1,5-a]pyrazin-4(5H)-one (2a). Yield 22.4 g (86%), yellow powder, mp 212–214°C. IR spectrum, ν, cm–1: 1681 (C=O), 3326 (NH). 1H NMR spectrum, δ, ppm: 7.19–7.22 (2H, m, H-3,6); 7.94 (1H, s, H-2); 11.43 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 69.5; 101.1; 123.1; 134.1; 139.9; 155.6. Mass spectrum, m/z (Irel, %): 262 [M+H]+ (100). Found, %: C 27.82; H 1.44; N 16.02. C6H4IN3O. Calculated, %: C 27.61; H 1.54; N 16.10.
7-Iodo-2-methylpyrazolo[1,5-a]pyrazin-4(5H)-one (2b). Yield 25.3 g (92%), light-brown powder, mp 222–224°C. IR spectrum, ν, cm–1: 1679 (C=O), 3330 (NH). 1H NMR spectrum, δ, ppm: 2.35 (3H, s, CH3); 6.99 (1H, s, H-6); 7.10 (1H, s, H-3); 11.31 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 13.9; 69.3; 106.2; 122.2; 134.6; 149.1; 155.3. Mass spectrum, m/z (Irel, %): 275 [M+H]+ (100). Found, %: C 30.36; H 2.31; N 15.07. C7H6IN3O. Calculated, %: C 30.57; H 2.20; N 15.28.
2-Ethyl-7-iodopyrazolo[1,5-a]pyrazin-4(5H)-one (2c). Yield 22.8 g (79%), yellow powder, mp 201–202°C. IR spectrum, ν, cm–1: 1677 (C=O), 3328 (NH). 1H NMR spectrum, δ, ppm (J, Hz): 1.24 (3H, t, J = 7.6, CH3); 2.73 (2H, q, J = 7.6, CH2); 7.04 (1H, s, H-3); 7.11 (1H, d, J = 5.2, H-6); 11.35 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 14.1; 21.4; 63.3; 104.8; 122.2; 134.5; 154.9; 155.3. Mass spectrum, m/z (Irel, %): 289 [M+H]+ (100). Found, %: C 33.02; H 2.93; N 14.44. C8H8IN3O Calculated, %: C 33.24; H 2.79; N 14.54.
7-Iodo-2-phenylpyrazolo[1,5-a]pyrazin-4(5H)-one (2d). Yield 28.3 g (84%), brown powder, mp 245–246°C. IR spectrum, ν, cm–1: 1680 (C=O), 3332 (NH). 1H NMR spectrum, δ, ppm (J, Hz): 7.20 (1H, s, H-6); 7.39–7.48 (3H, m, H Ph); 7.72 (1H, s, H-3); 7.98 (2H, d, J = 7.6, H Ph); 11.45 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 69.1; 103.2; 122.8; 125.8; 128.6; 128.8; 131.7; 135.0; 150.2; 155.0. Mass spectrum, m/z (Irel, %): 338 [M+H]+ (100). Found, %: C 42.89; H 2.24; N 12.35. C12H8IN3O. Calculated, %: C 42.75; H 2.39; N 12.46.
Synthesis of compounds 3a–d (General method). A solution of 7-iodopyrazolopyrazine 2a–d (0.02 mol), Et3N (4.44 g, 0.044 mol) and Pd(dppf)Cl2 (490 mg, 0.6 mmol) in MeOH (300 ml) was placed in a 1-l autoclave in which CO was pumped at 20 atm. The autoclave was kept at 100°C for 16 h, cooled to room temperature, and carefully ventilated for 0.5 h. The reaction mixture was filtered off, the filtrate was distilled off, H2O (200 ml) was added to the solid residue, the resulting precipitate was filtered off, washed with H2O (25 ml), MTBE (20 ml), and dried in air.
Methyl 4-oxo-4,5-dihydropyrazolo[1,5-a]pyrazine-7-carboxylate (3a). Yield 2.58 g (67%), gray powder, mp 218–220°C. IR spectrum, ν, cm–1: 1680, 1725 (C=O), 3346 (NH). 1H NMR spectrum, δ, ppm (J, Hz): 3.83 (3H, s, OCH3); 7.10 (1H, d, J = 2.0, H-3); 7.64 (1H, s, H-6); 7.98 (1H, d, J = 2.0, H-2); 11.81 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 52.3; 105.4; 112.8; 125.2; 133.7; 140.8; 155.3; 159.8. Mass spectrum, m/z (Irel, %): 194 [M+H]+ (100). Found, %: C 49.88; H 3.54; N 21.86. C8H7N3O3. Calculated, %: C 49.74; H 3.65; N 21.75.
Methyl 2-methyl-4-oxo-4,5-dihydropyrazolo[1,5-a]-pyrazine-7-carboxylate (3b). Yield 3.02 g (73%), gray powder, mp 221–223°C. IR spectrum, ν, cm–1: 1682, 1723 (C=O), 3344 (NH). 1H NMR spectrum, δ, ppm (J, Hz): 2.37 (3H, s, CH3); 3.84 (3H, s, OCH3); 6.88 (1H, s, H-3); 7.56 (1H, d, J = 4.8 H-6); 11.69 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 13.4; 52.1; 104.5; 112.6; 124.1; 134.3; 149.8; 154.9; 159.7. Mass spectrum, m/z (Irel, %): 208 [M+H]+ (100). Found, %: C 52.31; H 4.27; N 20.36. C9H9N3O3. Calculated, %: C 52.17; H 4.38; N 20.28.
Methyl 2-ethyl-4-oxo-4,5-dihydropyrazolo[1,5-a]-pyrazine-7-carboxylate (3c). Yield 3.71 g (84%), white powder, mp 196–198°C. IR spectrum, ν, cm–1: 1682, 1720 (C=O), 3342 (NH). 1H NMR spectrum, δ, ppm (J, Hz): 1.22 (1H, t, J = 7.6, CH3); 2.71 (2H, q, J = 7.6, CH2); 3.82 (3H, s, OCH3); 6.94 (1H, s, H-3); 7.58 (1H, s, H-6); 11.78 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 18.7; 26.2; 57.2; 108.3; 117.9; 129.2; 139.5; 160.1; 160.9; 164.9. Mass spectrum, m/z (Irel, %): 222 [M+H]+ (100). Found, %: C 54.41; H 5.15; N 18.93. C10H11N3O3. Calculated, %: C 54.30; H 5.01; N 19.00.
Methyl 4-oxo-2-phenyl-4,5-dihydropyrazolo[1,5-a]-pyrazine-7-carboxylate (3d). Yield 28.3 g (79%), brown powder, mp 244–246°C. IR spectrum, ν, cm–1: 1685, 1726 (C=O), 3347 (NH). 1H NMR spectrum, δ, ppm (J, Hz): 3.86 (3H, s, OCH3); 7.38 (1H, t, J = 7.2, H Ph); 7.47 (2H, t, J = 7.2, H Ph); 7.62 (1H, s, H-3); 7.69 (1H, s, H-6); 7.98 (2H, d, J = 7.2, H Ph); 12.15 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 52.3; 102.3; 112.7; 125.4; 126.0; 128.8; 129.0; 131.9; 135.4; 151.7; 155.2; 159.7. Mass spectrum, m/z (Irel, %): 270 [M+H]+ (95). Found, %: C 62.57; H 4.02; N 15.24. C14H11N3O3. Calculated, %: C 62.45; H 4.12; N 15.61.
Synthesis of compounds 4a–d (General method). Methyl carboxylate 3a–d (5 mmol) was added to a solution of KOH (295 mg, 5.25 mmol) in H2O (20 ml) and boiled for 8 h. The reaction mixture was cooled to room temperature, acidified with concentrated HCl to pH 3, the resulting precipitate was filtered off, washed with H2O (10 ml), and dried in air.
4-Oxo-4,5-dihydropyrazolo[1,5-a]pyrazine-7-carboxylicacid (4a). Yield 840 mg (94%), gray powder, mp 264–267°C. IR spectrum, ν, cm–1: 1678, 1687 (C=O), 3338 (NH). 1H NMR spectrum, δ, ppm (J, Hz): 7.11 (1H, d, J = 2.0, H-3); 7.64 (1H, s, H-6); 8.01 (1H, d, J = 2.0, H-2); 11.82 (1H, br. s, NH); 13.17 (1H, br. s, COOH). 13C NMR spectrum, δ, ppm: 105.7; 113.5; 125.3; 134.1; 140.8; 155.7; 160.8. Mass spectrum, m/z (Irel, %): 180 [M+H]+ (100). Found, %: C 46.85; H 2.73; N 23.61. C7H5N3O3. Calculated, %: C 46.93; H 2.81; N 23.46.
2-Methyl-4-oxo-4,5-dihydropyrazolo[1,5-a]pyrazine-7-carboxylic acid (4b). Yield 920 mg (95%), white powder, mp 245–247°C. IR spectrum, ν, cm–1: 1676, 1690 (C=O), 3335 (NH). 1H NMR spectrum, δ, ppm: 2.38 (3H, s, CH3); 6.90 (1H, s, H-3); 7.58 (1H, s, H-6); 11.77 (1H, br. s, NH); 13.21 (1H, br. s, COOH). 13C NMR spectrum, δ, ppm: 13.3; 104.4; 112.6; 124.1; 134.1; 149.5; 154.9; 160.3. Mass spectrum, m/z (Irel, %): 194 [M+H]+ (100). Found, %: C 49.85; H 3.58; N 21.66. C8H7N3O3. Calculated, %: C 49.74; H 3.65; N 21.75.
2-Ethyl-4-oxo-4,5-dihydropyrazolo[1,5-a]pyrazine-7-carboxylic acid (4c). Yield 960 mg (93%), light-brown powder, mp 224–226°C. IR spectrum, ν, cm–1: 1676, 1690 (C=O), 3331 (NH). 1H NMR spectrum, δ, ppm (J, Hz): 1.22 (3H, t, J = 6.9 CH3); 2.69–2.76 (2H, m, CH2); 6.93 (1H, s, H-3); 7.56 (1H, s, H-6); 11.79 (1H, br. s, NH). The proton of the COOH group is in exchange with the H2O of the deuterosolvent. 13C NMR spectrum, δ, ppm: 18.7; 26.0; 108.3; 117.8; 129.4; 139.3; 160.2; 160.5; 165.5. Mass spectrum, m/z (Irel, %): 208 [M+H]+ (100). Found, %: C 52.31; H 4.27; N 20.19. C9H9N3O3. Calculated, %: C 52.17; H 4.38; N 20.28.
4-Oxo-2-phenyl-4,5-dihydropyrazolo[1,5-a]pyrazine-7-carboxylic acid (4d). Yield 1.14 g (90%), brown powder, mp 290–293°C. IR spectrum, ν, cm–1: 1678, 1693 (C=O), 3331(NH). 1H NMR spectrum, δ, ppm (J, Hz): 7.37 (1H, t, J = 7.2, H Ph); 7.46 (2H, t, J = 7.2, H Ph); 7.61 (1H, s, H-3); 7.66 (1H, s, H-6); 7.97 (2H, d, J = 7.2, H Ph); 11.86 (1H, br. s, NH). The proton of the COOH group is in exchange with H2O of the deuterosolvent. 13C NMR spectrum, δ, ppm: 100.8; 111.6; 123.6; 124.6; 127.4; 127.5; 130.2;. 133.8; 150.0; 153.8;. 159.0. Mass spectrum, m/z (Irel, %): 256 [M+H]+ (95). Found, %: C 61.25; H 3.47; N 16.59. C13H9N3O3. Calculated, %: C 61.18; H 3.55; N 16.46.
Supplementary information file containing 1H and 13C NMR spectra of the synthesized compounds is available at the journal website http://springerlink.bibliotecabuap.elogim.com/journal/10593.
References
(a) Abd ul-Malic, M. A.; Zaki R. M.; El-Dean A. M. K.; Radwan, S. M. J. Heterocycl. Chem. 2018, 55, 1828. (b) Zaremba, O. V.; Gorobets, N. Yu.; Kovalenko, S. S.; Drushlyak, O. G.; Grevtsov, O. Yu.; Kovalenko, S. M. Chem. Heterocycl. Compd. 2013, 49, 915. (c) Liverton, N.; Kuduk, S. D.; Bechore, D. C.; Meng, N.; Luo, Y. WO Patent 2016101119A1. (d) Hrynyshyn, Ye. V.; Tsyzoryk, N. M.; Musiychuk, A. R.; Bol'but, A. V.; Vovk, M. V. Chem. Heterocycl. Compd. 2017, 53, 1242. (e) Hrynyshyn, Ye. V.; Musiychuk, A. R.; Tsizorik, N. M.; Bol'but, A. V.; Vovk, M. V. Chem. Heterocycl. Compd. 2019, 55, 1070. (f) Woll, M. G.; Qi, H.; Turpoff, A.; Zhang, N.; Zhang, X.; Chen, G.; Li, C.; Huang, S.; Yang, T.; Moon, Y.-C.; Lee, C.-S.; Choi, S.; Almstead, N. G.; Naryshkin, N. A.; Dakka, A.; Narasimhan, J.; Gabbeta, V.; Welch, E.; Zhao, X.; Richer, N.; Sheedy, J.; Weetall, M.; Karp, G. M. J. Med. Chem. 2016, 59, 6070. (g) Martinez Gonzalez, S.; Hernandez, A. I.; Alvarez, R. M.; Rodriguez, A.; Ramos-Lima, F.; Bischof, J. R.; Albarran, M. I.; Cebria, A.; Hernandez-Encinas, E.; Garcia-Arocha, J.; Cebrian, D.; Blanco-Aparicio, C.; Pastor, J. Bioorg. Med. Chem. Lett. 2017, 27, 4794. (h) Andrews, S. W.; Blake, J. F.; Haas, J.; Jiang, X.; Kolakowski, G. R.; Mareno, D. A.; Ren, L.; Walls, S. W. WO Patent 2018136661A1. (i) Allen, S.; Boys, M. L.; Chicarelli, M. J.; Fell, J. B.; Fisher, J. P.; Gaudino, J.; Hisken, E. J.; Hinklin, R. G.; Kraser, C. F.; Laird, E.; Robinson, J. E.; Tang, T. P.; Burgess, L. E.; Reiger, R. A.; Pheneger, J.; Saton, Y.; Leftheris, K.; Raneja, R. K.; Bennet, B. L. WO Patent 2016090385. (j) Di Fabio, R.; Gentile, G.; Pozzan, A.; Tarsi, L.; Terreni, S.; Tonelli, F. WO Patent 2009130232A1. (k) Arban, R.; Bianchi, F.; Buson, A.; Cremonesi, S.; Di Fabio, R.; Gentile, G.; Micheli, F.; Pasquarello, A.; Pozzan, A.; Tarsi, L.; Terreni, S.; Tonelli, F. Bioorg. Med. Chem. Lett. 2010, 20, 5044.
Askew, B. C.; Bednar, R. A.; Bednar, B.; Claremon, D. A.; Cook, J. J.; McIntyre, C. J.; Hunt, C. A.; Gould, R. J.; Lynch, R. J.; Lynch, J. J.; Gaul, S. L.; Strauieri, M. T.; Sitko, G. R.; Holahan, M. A.; Glass, J. D.; Hamill, T.; Gorham, L. M.; Prueksaritanont, T.; Baldwin, J. J.; Hartman, G. D. J. Med. Chem. 1997, 40, 1779.
Askew, B. C.; McIntyre, C. J.; Hunt, C. A.; Claremon, D. A.; Gould, R. J.; Lynch, R. J.; Armstrong, D. J. Bioorg. Med. Chem. Lett. 1995, 5, 475.
Sakurai, F.; Yukowa, T.; Kina, A.; Murakomi, M.; Takami, K.; Morimoto, S.; Seto, M.; Kamara, M.; Yamashita, T.; Nakashima, K.; Narita, N.; Bettini, E.; Ugolini, A.; Cotsi, M.; Hasui, T. Bioorg. Med. Chem. 2022, 56, 116576.
Conn, P. J.; Lindsley, G. W.; Stauffer, S. R.; Jones, C. K.; Conde-Ceide, S.; Tong, H. M.; Bartolome-Nebreda, J. M.; Macdonald, G. J. WO Patent 2012078817A1.
Surase, Y. B.; Kirandeep, S.; Amale, S. R.; Sood, R.; Purnapatre, K. P.; Pareek, P. K.; Das, B.; Nanda, K.; Kumar, S.; Verma, A. K. Bioorg. Med. Chem. Lett. 2017, 27, 3454.
Zhang, J.-H.; Fan, C.-D.; Zhao, B.-X.; Shin, D.-S.; Dong, W.-L.; Xie, Y.-S.; Miao, J.-Y. Bioorg. Med. Chem. 2008, 16, 10165.
Zheng, L-W.; Shao, J.-H.; Zhao, B.-X.; Miao, J.-Y. Bioorg. Med. Chem. Lett. 2011, 21, 3909.
Langford, H. M.; Williams, P. D.; Homnick, C. F.; Vacca, J. P.; Felock, P. J.; Stillmock, K. A.; Witmer, M. V.; Hasuda, D. J.; Gabryelski, L. J.; Schleif, W. A. Bioorg. Med. Chem. Lett. 2008, 18, 721.
Dorsch, D.; Buchtaller, H.-P.; Moinet, G.; Wegener, A. WO Patent 2013143663A1.
Tanaka, Y.; Ohashi, T.; Ikeda, Z.; Tanaka, Y.; Pünner, F.; Yamamoto, T.; Kakegawa, K.; Kikuchi, F.; Morishita, N.; Kasahara, T.; Seto, M.; Nakamura, M.; Takami, K.; Murakami, M.; Daini, M.; Mikami, S.; Sasaki, M. US Patent 20210087186A1.
Muller, S.; Schole-Loop, R.; Ortega, H. N.; Susmeier, F.; Jimenez, N. E.; Brumby, T.; Lindmer, N.; Gerder, C.; Pook, E.; Buchmuller, A.; Gaugaz, F. Z.; Lang, D.; Zimmerman, S.; Ehrmann, A. H. M.; Gerisch, M.; Lehmann, L.; Timmerman, A.; Schafer, M.; Schmidt, G.; Schlemmer, K.-H.; Follmann, M.; Kersten, E.; Wang, V.; Goo, X.; Wang, Y. WO Patent 2019, 219517.
Wu, G.; Albers, A.; Buell, J.; Burton, E. A.; Pham, P.; Powers, H.; Shi, S.; Spevak, W.; Wu, J.; Zhang, J. WO Patent 2018136202A2.
Board, J.; Wang, J.-X.; Crew, A. P.; Jin, M.; Foreman, K.; Mulvihill, M. J.; Sniechkus, V. Org. Lett. 2009, 11, 5118.
Bisballe, N.; Nedidi, M.; Demmer, C. S.; Chevallier, F.; Roisnel, T.; Dorset, V.; Halauko, Y. S.; Ivashkevich, O. A.; Matulis, V. E.; Bentabed-Ababsa, G.; Bunch, L.; Mongin, F. Eur. J. Org. Chem. 2018, 3904.
Korsik, M.; Tse, E. G.; Smith, D. G.; Lewis, W.; Rutledge, P. J.; Todd, M. H. J. Org. Chem. 2020, 85, 13438.
Brennführer, A.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2009, 48, 4114.
(a) Sidrauski, C.; Pliuschev, M.; Frost, J. M.; Black, L. A.; Xu, X.; Sweis, R. F.; Shi, L.; Zhang, Q. I.; Tong, Y.; Hutchins, C. W.; Chung, S.; Dart, M. J. WO Patent 2017193063A1. (b) Martin, K.; Sidrauski, C.; Frost, J.; Pliuschev, M.; Tong, Y.; Black, L.; Xu, X.; Shi, L.; Zhang, Q.; Chung, S.; Sweis, R.; Dart, M.; Randolph, J.; Murauski, K. WO Patent 2019090076A1. (c) Tsizorik, N. M.; Hrynyshyn, Ye. V.; Musiychuk, A. R.; Bol'but, A. V.; Nechayev, M. A.; Vovk, M. V. Chem. Heterocycl. Compd. 2020, 56, 1554.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Geterotsiklicheskikh Soedinenii, 2023, 59(6/7), 425–428
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tsyzoryk, N.M., Bol’but, A.V., Loza, K.I. et al. A convenient synthesis of methyl 4-oxo-4,5-dihydropyrazolo[1,5-a]-pyrazine-7-carboxylates and corresponding carboxylic acids. Chem Heterocycl Comp 59, 425–428 (2023). https://doi.org/10.1007/s10593-023-03212-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-023-03212-z