

N-Boc-protected 5-formyl-1H-pyrazol-4-amines react with malonic acid in pyridine in the presence of pyrrolidine at 45–50°С or with malonic acid monomethyl ether in the presence of pyrrolidine in AcOH under reflux with the formation of 5-oxo-4,5-dihydro-1Hpyrazolo[4,3-b]pyridine-6-carboxylic acids. The reaction of N-Boc-protected 5-formyl-1H-pyrazol-4-amines with cyanoacetic acid in pyridine in the presence of pyrrolidine at 45–50°С leads to the formation of 5-oxo-4,5-dihydro-1H-pyrazolo[4,3-b]pyridine-6-carbonitriles. The latter can also be obtained via cyclocondensation of N-Boc-protected 5-formyl-1H-pyrazol-4-amines with methyl cyanoacetate in AcOH under reflux in the presence of pyrrolidine or in MeCN containing pyrrolidine and a catalytic amount of proline heated under reflux.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Derivatives of 5-oxo-4,5-dihydro-1H-pyrazolo[4,3-b]- pyridine are condensed nitrogen-containing heterocyclic compounds with pronounced biological activity. Among them, inhibitors of p38 mitogen-activated protein kinases, promising agents for the treatment of viral diseases, arthritis, Alzheimer's disease and dermatitis,1 inhibitors of bromodomain and extraterminal proteins for the treatment of autoimmune diseases, viral and inflammatory infections,2 as well as NIK kinase inhibitors effective for the treatment and prevention of inflammatory diseases3 were identified. There is also a report on the use of derivatives of 5-oxo-4,5-dihydro-1H-pyrazolo[4,3-b]-pyridine as important intermediates in the synthesis of chemokine receptor antagonists pyrazolopiperidines.4
However, despite the fairly wide pharmacological profile of 5-oxo-4,5-dihydro-1H-pyrazolo[4,3-b]pyridine derivatives, the known methods for their synthesis are multistage, require the use of expensive and aggressive reagents, and, as a rule, include a series of successive conversions to annulate the pyridine ring to the pyrazole.1b, 3,5 In particular, the condensation of 3,5-diacyl-1-phenyl-1H-pyrazol-4-amines with cyanoacetic acid proceeds in Ac2O medium under the conditions of microwave activation with the formation of 2-cyano-N-(3,5-diacyl-1-phenyl-1H-pyrazol-4-yl)acetamides, which cyclize into 5-oxo-4,5-dihydro-1H-pyrazolo[4,3-b]pyridine-6-carbonitriles only upon heating in DMF under reflux in the presence of NaOAc.5a In turn, intramolecular copper-catalyzed amidation of 4-iodopyrazoles does not exhibit high selectivity and leads to the formation of 5-oxo-4,5-dihydro-1H-pyrazolo[4,3-b]pyridine derivatives with relatively low yields.5b In the context of the foregoing, it seems reasonable to develop a preparatively convenient way to access new representatives of this heterocyclic system, additionally containing a carboxyl or nitrile functionality in the pyridine ring, which can be effectively subjected to structural modifications by pharmacophore groups. The choice of the indicated functional groups as substituents is also due to the powerful synthetic potential6 and biological activity7 of isostere 3-carbo-functionalized quinolin-2-ones.
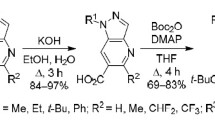
In a previous report, we showed that the Friedlander reaction of N-Boc-protected 1-alkyl- or 1-aryl-5-formyl-1Hpyrazol-4-amines with various methylene-containing ketones can be used to obtain 5-substituted pyrazolo[4,3-b]-pyridines.8 Considering these revelations, as well as the literature data on the cyclocondensation of aromatic o-aminoaldehydes or ketones with CH acids as an effective method for the synthesis of 3-substituted quinolones,6a,7a,b,9 we investigated the reactions of N-Boc-protected 1-alkyl- or 1-aryl-5-formyl-1H-pyrazol-4-amines 1a–g with malonic acid (2a), its methyl ester 2b, and also cyanoacetic acid (2c) and its methyl ester 2d in this work.
In search of the optimal conditions for cyclocondensation of 5-formyl-1H-pyrazol-4-amines 1a–g with malonic acid (2a), it was found that the use of pyridine as the solvent and an equimolar amount of pyrrolidine as the catalyst is the most effective. In this medium, the reaction is complete at 45–50°С within 8–12 h and proceeds via the intermediate products 3a–g, which can be observed by 1H NMR spectroscopy (Table 1, method I). Subsequent treatment with 2 N aqueous HCl removes the N-Boc protection to effect cyclization to the desired 5-oxo-4,5-dihydro-1Hpyrazolo[4,3-b]pyridine-6-carboxylic acids 4a–g in 54–86% yields. Carrying out the reaction under reflux in the AcOH–pyrrolidine system, which was previously used to condense N-Boc-protected 1-alkyl- or 1-aryl-5-formyl-1H-pyrazol-4-amines with ketones,8 was ineffective. Thus, after carrying out the reaction of 5-formyl-1H-pyrazol-4-amine 1a with acid 2a under these conditions, it was found that the content of the target product 4a in the reaction mixture was only 18% according to LC-M spectrometry. However, the replacement of malonic acid (2a) with its monomethyl ether 2b allowed to successfully effect cyclocondensation with N-Boc-protected 1-alkyl- or 1-aryl-5-formyl-1Hpyrazol-4-amines 1a–e by stirring in AcOH–pyrrolidine at room temperature for 3 h, followed by heating under reflux for 5–6 h (method II). Most probably that under these conditions the cyclization stage proceeds noticeably faster than the competing decarboxylation reaction of the malonate carboxyl group of the crotonic condensation intermediate, although the target compounds 4a–e are obtained in 4–15% lower yields than when using method I. Formation of the pyridine ring as a result of the studied cyclization is confirmed by the presence in the IR spectra of compounds 4a–g of absorption bands of N–H in the 3201–3217 cm–1 range, carboxylic C=O in the 1721–1739 cm–1 range, pyridone C=O in the 1652–1665 cm–1 range, whereas the 1H NMR spectra contained the singlet signals of protons of 7-CH at 8.70–8.97 (for compounds 4a–d,f,g) and 9.63 ppm (for compound 4e), of NH at 13.26–13.61 ppm, and of СО2Н at 15.20–15.50 ppm.
The reactivity of cyanoacetic (compound 2c) and malonic acid (compound 2a) with 5-formyl-1H-pyrazol-4-amines 1a,c,e,g are practically the same. 5-Oxo-4,5-dihydro-1H-pyrazolo[4,3-b]pyridine-6-carbonitriles 1a,c,e,g are formed as a result of this reaction in the pyridine–pyrrolidine system at 45–50°С in 48–74% yields (Table 2, method I). At the same time, methyl cyanoacetate (2d) does not react with 5-formyl-1H-pyrazol-4-amines 1a–g under these conditions. A positive result, however, was achieved by carrying out the reaction in AcOH under reflux in the presence of an equimolar amount of pyrrolidine (method II).
The use of the pyrrolidine–proline catalytic system, which proved to be effective when using MeCN as a solvent, is also noteworthy. Using the reaction of bifunctional pyrazoles 1a–d with ester 2d in MeCN under reflux as an example, it was demonstrated that the use of an equimolar amount of pyrrolidine and 0.1 equiv of proline leads to the desired products 5a–d in 52–72% yields (Table 2, method III). It should be noted that the yields of compounds 5a–d decrease by 8–11% in the absence of proline.
In the case of method III, the reaction most likely proceeds through the formation of Knoevenagel product 6, detected by LC-M spectrometry. Subsequently, intramolecular acylation of intermediate 6 occurs by the action of pyrrolidine as a base, with the formation of intermediate 7.10N-Boc protection is removed under the reaction conditions (elevated temperature, the presence of a base and formed H2O), which is consistent with published data11 (Scheme 1).

Scheme 1
Replacing the carboxyl group in acids 4a–g with a cyano group affects the spectral characteristics of nitriles 5a–g. Thus, in the IR spectra of products 5a–g, an insignificant short-wavelength shift of the absorption bands of N–H bonds (3205–3252 cm–1), a short-wavelength shift of the absorption bands of C=O bonds of pyridone (1658–1694 cm–1), as well as the appearance of medium intensity absorption bands of С≡N groups in the 2220–2230 cm–1 range are observed. In the 1H NMR spectra of nitriles 5a–g, the chemical shifts of the 7-CH proton singlet signals are practically unchanged; however, the NH proton signal does undergo a noticeable upfield shift of 1 ppm.
To conclude, we have proposed a simple and convenient method for the synthesis of promising novel synthetic intermediates, 5-oxo-4,5-dihydro-1H-pyrazolo[4,3-b]pyridine-6-carboxylic acids and their nitriles. This approach is based on the cyclocondensation of N-Boc-protected 1-alkyl- or 1-aryl-5-formyl-1H-pyrazol-4 amines with malonic or cyanoacetic acids and their esters.
Experimental
IR spectra were registered on a Bruker Vertex 70 FT-IR spectrometer in KBr pellets. 1H and 13C NMR spectra were acquired on a Varian VXR-400 spectrometer (400 and 126 MHz, respectively) in CDCl3 (compounds 1b,f,g and 9b,f,g) or DMSO-d6 (compounds 4a–g and 5a–g), with TMS as internal standard. Mass spectra were recorded on a Agilent LC/MSD SL LC-MS system equipped with a Zorbax SB-C18 column (4.6 × 150 mm, 1.8 μm), atmospheric pressure electrospray ionization, DMSO solvent. Elemental analysis was performed on a PerkinElmer Series II 2400 Elemental analyzer. Melting points were determined on a Kofler bench and are uncorrected.
The starting pyrazoles 1а,121с–е,8 and 8b,f,g8 were obtained according to published methods. 1-Ethyl-1Н-pyrazol-4-amine, 3-methoxy-1-methyl-1Н-pyrazol-4-amine, and 1-methyl-3-phenyl-1Н-pyrazol-4-amine used in the synthesis of N-Boc-protected 1-alkyl-5-formyl-1H-pyrazol-4-amines 1b,f,g were supplied by Enamine Ltd.
Synthesis ofN-Boc-substituted 1-alkyl-1H-pyrazol-4-amines 8b,f,g (General method).8 DMAP (1.22 g, 0.01 mol) was added with stirring and cooling to 0°С to a solution of pyrazol-4-amine (0.257 mol) in THF (400 ml), followed by di-tert-butyl dicarbonate (56.8 g, 0.260 mol) over 20 min. The reaction mixture was stirred at 0°C for 1 h, then at room temperature for 4–6 h, and finally at 40°С for 1 h. The solvent was evaporated under reduced pressure, the residue was extracted with MTBE (300 ml). MTBE–hexane, 3:2 mixture (300 ml) was added to the resulting oily product. The precipitate formed after stirring was filtered off and air-dried.
tert-Butyl (1-ethyl-1Н-pyrazol-4-yl)carbamate (8b) was obtained from 1-ethyl-1Н-pyrazol-4-amine (35.4 g, 0.319 mol). Yield 61.3 g (91%), light-pink powder, mp 68–69°C. IR spectrum, ν, cm–1: 3376 (N–H), 1725 (С=О). 1H NMR spectrum, δ, ppm (J, Hz): 1.41–1.48 (12H, m, CH3, С(CH3)3); 4.08 (2H, q, J = 7.2, CH2); 6.40 (1H, br. s, NH); 7.28 (1H, s, H-5); 7.65 (1H, s, Н-3). 13C NMR spectrum, δ, ppm: 14.9; 27.8; 46.7; 79.5; 118.9; 121.1; 129.2; 152.8. Mass spectrum, m/z (Irel, %): 212 [М+Н]+ (100). Found, %: C 56.39; H 8.20; N 19.68. C10H17N3O2. Calculated, %: C 56.85; H 8.11; N 19.89.
tert-Butyl (3-methoxy-1-methyl-1Н-pyrazol-4-yl)carbamate (8f) was obtained from 3-methoxy-1-methyl-1Н-pyrazol-4-amine (24.9 g, 0.196 mol). Yield 42.3 g (95%), colorless crystals, mp 69–70°C. IR spectrum, ν, cm–1: 3285 (N–H), 1714 (С=О). 1H NMR spectrum, δ, ppm: 1.47 (9H, s, С(CH3)3); 3.66 (3H, s, NCH3); 3.89 (3H, s, ОCH3); 6.08 (1H, br. s, NH); 7.48 (1H, s, H-5). 13C NMR spectrum, δ, ppm: 28.3; 38.9; 56.3; 80.1; 105.7; 123.1; 153.1; 153.7. Mass spectrum, m/z (Irel, %): 228 [М+Н]+ (100). Found, %: C 53.10; H 7.61; N 18.31. C10H17N3O3. Calculated, %: C 52.85; H 7.54; N 18.49.
tert-Butyl (1-methyl-3-phenyl-1Н-pyrazol-4-yl)carbamate (8g) was obtained from 1-methyl-3-phenyl-1Н-pyrazol-4-amine (24.6 g, 0.142 mol). Yield 34.2 g (88%), brown powder, mp 93–94°C. IR spectrum, ν, cm–1: 3301 (N–H), 1687 (С=О). 1H NMR spectrum, δ, ppm (J, Hz): 1.12 (9H, s, С(CH3)3); 3.89 (3H, s, NCH3); 7.25–7.40 (4H, m, NH, H-3,4,5 Ph); 7.67 (2H, d, J = 7.2, H-2,6 Ph); 7.80 (1H, s, H-5). 13C NMR spectrum, δ, ppm: 27.8; 38.8; 80.0; 117.6; 122.7; 127.3; 128.5; 132.1; 140.8; 152.8. Mass spectrum, m/z (Irel, %): 274 [М+Н]+ (100). Found, %: C 65.68; H 7.08;N 15.49. C15H19N3O2. Calculated, %: C 65.91; H 7.01; N 15.37.
Synthesis ofN-Boc-protected 1-alkyl-5-formyl-1Hpyrazol-4-amines 1b,f,g (General method).8 n-BuLi (2.5 M solution in hexane, 159 ml, 0.40 mol) was added to a cooled (–78°С) solution of carbamate 8b,f,g (0.18 mol) in THF (600 ml) under argon atmosphere. The reaction mixture was stirred at –78°С for 2–3 h. DMF (15.4 ml, 0.20 mol) was added to the reaction mixture at –78°C, then stirring was continued at room temperature for 10–12 h. Saturated aqueous NH4Cl (200 ml) was added to the mixture. The formed mixture was stirred for additional 20–30 min and extracted with MTBE (3×200 ml). The organic layer was washed with saturated aqueous NaCl (2×150 ml), dried over anhydrous Na2SO4, and filtered through a layer of silica gel. The solvent was evaporated, MTBE–hexane, 3:2 mixture (150 ml) was added to the oily residue. The precipitate formed after stirring was filtered off and air-dried.
tert-Butyl (1-ethyl-5-formyl-1Н-pyrazol-4-yl)carbamate (1b). Yield 37.4 g (87%), light-pink powder, mp 68–69°C. IR spectrum, ν, cm–1: 3376 (N–H), 1725 (С=О aldehyde), 1698 (С=О carbamate). 1H NMR spectrum, δ, ppm (J, Hz): 1.47–1.50 (12H, m, CH3, С(CH3)3); 4.39 (2H, q, J = 7.2, CH2); 8.01 (1H, br. s, NH); 8.28 (1H, s, H-5); 9.98 (1H, s, СНО). 13C NMR spectrum, δ, ppm: 15.5; 27.7; 45.2; 80.7; 125.0; 128.2; 129.0; 152.0; 179.0. Mass spectrum, m/z (Irel, %): 240 [М+Н]+ (100). Found, %: C 55.38; H 7.21; N 17.49. C11H17N3O3. Calculated, %: C 55.22; H 7.16; N 17.56.
tert-Butyl (5-formyl-3-methoxy-1-methyl-1Н-pyrazol-4-yl)carbamate (1f). Yield 39.0 g (85%), colorless crystals, mp 76–77°C. IR spectrum, ν, cm–1: 3239 (N–H), 1710 (С=О aldehyde), 1678 (С=О carbamate). 1H NMR spectrum, δ, ppm: 1.49 (9H, s, С(CH3)3); 3.94 (3H, s, NCH3); 3.98 (3H, s, OCH3); 6.05 (1H, br. s, NH); 9.90 (1H, s, СНО). 13C NMR spectrum, δ, ppm: 27.6; 38.7; 56.1; 80.7; 109.5; 131.5; 153.6; 154.2; 180.6. Mass spectrum, m/z (Irel, %): 256 [М+Н]+ (100). Found, %: C 51.05; H 6.74; N 16.18. C11H17N3O4. Calculated, %: C 51.76; H 6.71; N 16.46.
tert-Butyl (5-formyl-1-methyl-3-phenyl-1Н-pyrazol-4-yl)carbamate (1g). Yield 31.4 g (58%), brown powder, mp 105–106°C. IR spectrum, ν, cm–1: 3258 (N–H), 1759 (С=О aldehyde), 1671 (С=О carbamate). 1H NMR spectrum, δ, ppm (J, Hz): 1.27 (9H, s, С(CH3)3); 3.94 (3H, s, NCH3); 7.14–7.23 (3H, m, H-3,4,5 Ph); 7.50 (2H, d, J = 7.6, H-2,6 Ph); 7.67 (1H, br. s, NH); 9.65 (1H, s, СНО). 13C NMR spectrum, δ, ppm: 27.6; 39.6; 80.1; 126.4; 127.5; 127.9; 131.1; 133.0; 144.2; 154.1; 179.7. Mass spectrum, m/z (Irel, %): 302 [М+Н]+ (100). Found, %: C 63.58; H 6.40; N 13.78. C16H19N3O3. Calculated, %: C 63.77; H 6.36; N 13.94.
Synthesis of pyrazolo[4,3-b]pyridine-6-carboxylic acids 4a–g (General method). Method I. Malonic acid (2a) (0.62 g, 6 mmol) followed by pyrrolidine (0.5 ml, 6 mmol) was added to a solution of N-Boc-protected 1-alkyl- or 1-aryl-5-formyl-1H-pyrazol-4-amine 1а–g (5 mmol) in pyridine (15 ml). The reaction mixture was heated at 45–50°С for 8–12 h. Pyridine was evaporated under reduced pressure, and the residue was treated with 2 N aqueous HCl (10 ml). The formed precipitate was filtered off, washed with H2O (10 ml) and MTBE (10 ml), air-dried, and recrystallized from MeOH.
Method IІ. Monomethyl malonate (2b) (0.70 g, 6 mmol) followed by pyrrolidine (0.5 ml, 6 mmol) was added to a solution of N-Boc-protected 1-alkyl- or 1-aryl-5-formyl-1Hpyrazol-4-amine (5 mmol) 1а–e (5 mmol) in glacial AcOH (20 ml). The reaction mixture was stirred at room temperature for 3 h, then heated under reflux for 5–6 h. The solvent was evaporated under reduced pressure, H2O (30–40 ml) was added to the oily residue, and stirred until a loose precipitate formed. It was filtered off, washed with H2O (10 ml) and MTBE (10 ml), air-dried, and recrystallized from MeOH.
1-Methyl-5-oxo-4,5-dihydro-1Н-pyrazolo[4,3-b]pyridine-6-carboxylic acid (4а). Yield 0.83 g (86%, method I), 0.69 g (71%, method IІ), light-yellow powder, mp 264–265°C. IR spectrum, ν, cm–1: 3201 (N–H), 1739 (C=О carboxylic acid), 1652 (C=О pyridone). 1H NMR spectrum, δ, ppm: 4.10 (3H, s, CH3); 7.69 (1H, s, H-3); 8.92 (1H, s, 7-CН); 13.26 (1H, s, NH); 15.38 (1H, s, CO2H). 13C NMR spectrum, δ, ppm: 37.2; 115.2; 124.0; 127.3; 129.2; 131.4; 163.9; 165.6. Mass spectrum, m/z (Irel, %): 194 [M+H]+ (100). Found, %: C 49.95; H 3.57; N 21.91. C8H7N3O3. Calculated, %: C 49.74; H 3.65; N 21.75.
1-Ethyl-5-oxo-4,5-dihydro-1Н-pyrazolo[4,3-b]pyridine-6-carboxylic acid (4b). Yield 0.85 g (82%, method I), 0.70 g (68%, method IІ), yellow powder, mp 259–261°C. IR spectrum, ν, cm–1: 3214 (N–H), 1732 (C=О carboxylic acid), 1663 (C=О pyridone). 1H NMR spectrum, δ, ppm (J, Hz): 1.39 (3H, t, J = 7.2, CH3); 4.50 (2H, q, J = 6.8, CH2); 7.74 (1H, s, H-3); 8.97 (1H, s, 7-CH); 13.27 (1H, s, NH); 15.39 (1H, s, CO2H). 13C NMR spectrum, δ, ppm: 15.6; 44.9; 115.3; 124.3; 126.5; 129.2; 131.2; 164.0; 165.6. Mass spectrum, m/z (Irel, %): 208 [M+H]+ (100). Found, %: C 52.32; H 4.23; N 20.46. C9H9N3O3. Calculated, %: C 52.17; H 4.38; N 20.28.
1-tert-Butyl-5-oxo-4,5-dihydro-1Н-pyrazolo[4,3-b]-pyridine-6-carboxylic acid (4с). Yield 0.73 g (62%, method I), 0.68 g (58%, method IІ), beige powder, mp 261–263°C. IR spectrum, ν, cm–1: 3212 (N–H), 1725 (C=О carboxylic acid), 1660 (C=О pyridone). 1H NMR spectrum, δ, ppm: 1.69 (9H, s, С(CH3)3); 7.75 (1H, s, H-3); 8.87 (1H, s, 7-CH); 13.38 (1H, s, NH); 15.39 (1H, s, CO2H). 13C NMR spectrum, δ, ppm: 30.1; 62.2; 114.7; 123.4; 125.2; 130.9; 132.2; 163.3; 165.6. Mass spectrum, m/z (Irel, %): 236 [M+H]+ (100). Found, %: C 55.93; H 5.65; N 17.75. C11H13N3O3. Calculated, %: C 56.16; H 5.57; N 17.86.
5-Oxo-1-phenyl-4,5-dihydro-1Н-pyrazolo[4,3-b]pyridine-6-carboxylic acid (4d). Yield 0.74 g (59%, method I), 0.66 g (52%, method IІ), brown powder, mp 269–272°C. IR spectrum, ν, cm–1: 3213 (N–H), 1729 (C=О carboxylic acid), 1658 (C=О pyridone). 1H NMR spectrum, δ, ppm (J, Hz): 7.52 (1H, t, J = 7.2, H-4 Ph); 7.65 (2H, t, J = 7.6, H-3,5 Ph); 7.76 (2H, d, J = 7.2, H-2,6 Ph); 8.07 (1H, s, H-3); 8.70 (1H, s, 7-CH); 13.53 (1H, s, NH); 15.20 (1H, s, CO2H). 13C NMR spectrum, δ, ppm: 116.5; 123.1; 125.8; 127.1; 128.5; 130.4; 130.8; 131.0; 138.6; 163.2; 165.3. Mass spectrum, m/z (Irel, %): 256 [M+H]+ (100). Found, %: C 60.98; H 3.58; N 16.33. C13H9N3O3. Calculated, %: C 61.18; H 3.55; N 16.46.
5-Oxo-1-(pyridin-2-yl)-4,5-dihydro-1Н-pyrazolo[4,3-b]-pyridine-6-carboxylic acid (4e). Yield 0.75 g (60%, method I), 0.61 g (48%, method IІ), brown powder, mp 284–287°C. IR spectrum, ν, cm–1: 3217 (N–H), 1724 (C=О carboxylic acid), 1656 (C=О pyridone). 1H NMR spectrum, δ, ppm (J, Hz): 7.44 (1H, t, J = 7.4, Н-5 Ру); 7.89 (1H, d, J = 6.2, Н-3 Ру); 8.08 (1H, t, J = 5.4, Н-4 Ру); 8.14 (1H, s, H-3); 8.63 (1Н, d, J = 5.4, Н-6 Ру); 9.63 (1H, s, 7-CH); 13.61 (1H, s, NH); 15.24 (1H, s, CO2H). 13C NMR spectrum, δ, ppm: 113.1; 117.0; 122.5; 125.4; 128.8; 131.9; 134.4; 140.1; 148.5; 152.5; 163.8; 165.3. Mass spectrum, m/z (Irel, %): 257 [M+H]+ (100). Found, %: C 56.16; H 3.22; N 21.75. C12H8N4O. Calculated, %: C 56.25; H 3.15; N 21.87.
3-Methoxy-1-methyl-5-oxo-4,5-dihydro-1Н-pyrazolo-[4,3-b]pyridine-6-carboxylic acid (4f). Yield 0.69 g (62%, method I), white powder, mp 264–265°C. IR spectrum, ν, cm–1: 3211 (N–H), 1725 (C=О carboxylic acid), 1665 (C=О pyridone). 1H NMR spectrum, δ, ppm: 3.94 (3H, s, NCH3); 3.97 (3H, s, OCH3); 8.85 (1H, s, 7-CH); 13.42 (1H, s, NH); 15.50 (1H, s, CO2H). 13C NMR spectrum, δ, ppm: 36.6; 57.3; 114.2; 116.5; 128.6; 131.5; 148.1; 163.7; 165.6. Mass spectrum, m/z (Irel, %): 224 [M+H]+ (100). Found, %: C 48.24; H 4.01; N 18.78. C9H9N3O4. Calculated, %: C 48.43; H 4.06; N 18.83.
1-Methyl-5-oxo-3-phenyl-4,5-dihydro-1Н-pyrazolo-[4,3-b]pyridine-6-carboxylic acid (4g). Yield 0.73 g (54%, method I), brown powder, mp 290–293°C. IR spectrum, ν, cm–1: 3216 (N–H), 1721 (C=О carboxylic acid), 1662 (C=О pyridone). 1H NMR spectrum, δ, ppm: 4.16 (3H, s, NCH3); 7.36–7.56 (3H, m, H-3,4,5 Ph); 7.75–7.95 (2H, m, H-2,6 Ph); 8.95 (1H, s, 7-CH); 13.28 (1H, s, NH); 15.25 (1H, s, CO2H). 13C NMR spectrum, δ, ppm: 38.8; 116.9; 128.1; 129.6; 130.8; 132.5; 133.0; 133.8; 136.6; 141.6; 166.2; 171.9. Mass spectrum, m/z (Irel, %): 270 [M+H]+ (100). Found, %: C 62.29; H 4.06; N 15.55. C14H11N3O3. Calculated, %: C 62.45; H 4.12; N 15.61.
Synthesis of pyrazolo[4,3-b]pyridine-6-carbonitriles 5a–g (General method). Method I. Compounds 5a,c,e,g were obtained by method I of the synthesis of compounds 4a–g from N-Boc-protected 1-alkyl- or 1-aryl-5-formyl-1Hpyrazol-4-amines 1a,c,e,g (5 mmol) and cyanoacetic acid (2c) (0.51 g, 6 mmol).
Method IІ. Methyl cyanoacetate (2d) (0.5 ml, 6 mmol) followed by pyrrolidine (0.5 ml, 6 mmol) was added to a solution of N-Boc-protected 1-alkyl- or 1-aryl-5-formyl-1Hpyrazol- 4-amine 1а–g (5 mmol) in AcOH (20 ml). The reaction mixture was stirred at room temperature for 2 h, then heated under reflux for 5–6 h. The solvent was evaporated under reduced pressure. H2O (50 ml) was added to the residue, and the mixture was stirred. The formed precipitate was filtered off, washed with H2O (10 ml) and MTBE (10 ml), air-dried, and recrystallized from MeOH.
Method IІІ. Methyl cyanoacetate (2d) (0.5 ml, 6 mmol) followed by pyrrolidine (0.5 ml, 6 mmol) and proline (6 mg, 0.5 mmol) was added to a solution of N-Bocprotected 1-alkyl- or 1-aryl-5-formyl-1H-pyrazol-4-amine 1а–d (5 mmol) in anhydrous MeCN (30 ml). The reaction mixture was stirred at room temperature for 2 h, then heated under reflux for 4–6 h. The solvent was evaporated under reduced pressure. H2O (20 ml) was added to the residue, and the mixture was stirred. The formed precipitate was filtered off, washed with H2O (10 ml) and MTBE (10 ml), air-dried, and recrystallized from MeOH.
1-Methyl-5-oxo-4,5-dihydro-1Н-pyrazolo[4,3-b]pyridine-6-carbonitrile (5а). Yield 0.64 g (74%, method I), 0.58 g (67%, method II), 0.48 g (56%, method III), yellow powder, mp 243–245°C. IR spectrum, ν, cm–1: 3251 (N–H), 2226 (C≡N), 1671 (C=O). 1H NMR spectrum, δ, ppm: 3.98 (3H, s, NCH3); 7.52 (1H, s, H-3); 8.81 (1H, s, 7-CH); 12.32 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 39.9; 103.4; 117.1; 123.7; 125.5; 130.1; 134.6; 159.2. Mass spectrum, m/z (Irel, %): 175 [M+H]+ (100). Found, %: C 55.36; H 3.57; N 32.32. C8H6N4O. Calculated, %: C 55.17; H 3.47; N 32.17.
1-Ethyl-5-oxo-4,5-dihydro-1Н-pyrazolo[4,3-b]pyridine-6-carbonitrile (5b). Yield 0.65 g (69%, method IІ), 0.49 g (52%, method IІІ), beige powder, mp 251–253°C. IR spectrum, ν, cm–1: 3232 (N–H), 2220 (C≡N), 1694 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 1.38 (3H, t, J = 7.2, CH3); 4.36 (2H, q, J = 6.8, CH2); 7.54 (1H, s, H-3); 8.83 (1H, s, 7-CH); 12.25 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 15.0; 44.4; 103.0; 116.7; 123.5; 124.1; 129.7; 134.1; 158.8. Mass spectrum, m/z (Irel, %): 189 [M+H]+ (100). Found, %: C 57.28; H 4.31; N 29.66. C9H8N4O. Calculated, %: C 57.44; H 4.29; N 29.77.
1-tert-Butyl-5-oxo-4,5-dihydro-1Н-pyrazolo[4,3-b]-pyridine-6-carbonitrile (5с). Yield 0.61 g (56%, method I), 0.73 g (67%, method IІ), 0.65 g (60%, method IІІ), brown powder, mp 279–281°C. IR spectrum, ν, cm–1: 3244 (N–H), 2228 (C≡N), 1674 (C=O). 1H NMR spectrum, δ, ppm: 1.64 (9H, s, С(CH3)3); 7.53 (1H, s, H-3); 8.97 (1H, s, 7-CH); 12.38 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 29.9; 61.6; 117.1; 122.8; 123.1; 131.7; 136.1; 158.7; 164.6. Mass spectrum, m/z (Irel, %): 217 [M+H]+ (100). Found, %: C 61.03; H 5.46; N 25.75. C11H12N4O. Calculated, %: C 61.10; H 5.59; N 25.91.
5-Oxo-1-phenyl-4,5-dihydro-1Н-pyrazolo[4,3-b]pyridine- 6-carbonitrile (5d). Yield 0.87 g (74%, method IІ), 0.85 g (72%, method IІІ), beige powder, mp 259–261°C. IR spectrum, ν, cm–1: 3205 (N–H), 2230 (C≡N), 1662 (C=О). 1H NMR spectrum, δ, ppm (J, Hz): 7.45 (1H, t, J = 7.2, H-4 Ph); 7.57 (2H, t, J = 7.6, H-3,5 Ph); 7.71 (2H, d, J = 7.2, H-2,6 Ph); 7.88 (1H, s, H-3); 8.78 (1H, s, 7-CH); 12.90 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 104.8; 116.9; 122.8; 124.0; 127.2; 128.3; 130.3; 132.4; 135.2; 138.8; 159.3. Mass spectrum, m/z (Irel, %): 237 [M+H]+ (100). Found, %: C 65.97; H 3.45; N 23.56. C13H8N4O. Calculated, %: C 66.10; H 3.41; N 23.72.
5-Oxo-1-(pyridin-2-yl)-4,5-dihydro-1Н-pyrazolo[4,3-b]-pyridine-6-carbonitrile (5е). Yield 0.57 g (48%, method I), 0.83 g (70%, method IІ), pale-yellow powder, mp 286–287°C. IR spectrum, ν, cm–1: 3247 (N–H), 2229 (C≡N), 1676 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 7.41 (1H, t, J = 7.1, H-5 Ру); 7.89 (1H, d, J = 6.0, H-3 Ру); 8.04 (1H, s, H-3); 8.06 (1H, t, J = 5.2, H-4 Ру); 8.56 (1Н, d, J = 5.2, H-6 Ру); 9.34 (1H, s, 7-CH); 13.50 (1H, s, NH). 13C NMR spectrum, δ, ppm: 105.0; 112.9; 116.7; 122.4; 123.4; 128.5; 132.7; 137.5; 140.1; 145.5; 152.2; 159.0. Mass spectrum, m/z (Irel, %): 238 [M+H]+ (100). Found, %: C 60.55; H 2.85; N 29.43. C12H7N5O. Calculated, %: C 60.76; H 2.97; N 29.52.
3-Methoxy-1-methyl-5-oxo-4,5-dihydro-1Н-pyrazolo-[4,3-b]pyridine-6-carbonitrile (5f). Yield 0.69 g (68%, method IІ), light-beige powder, mp 252–254°C. IR spectrum, ν, cm–1: 3224 (N–H), 2224 (C≡N), 1658 (C=О). 1H NMR spectrum, δ, ppm: 3.82 (3H, s, NCH3); 3.92 (3H, s, ОCH3); 8.73 (1H, s, 7-CH); 12.30 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 36.5; 57.2; 114.7; 117.1; 127.3; 134.4; 148.8; 152.6; 159.0. Mass spectrum, m/z (Irel, %): 204 [M+H]+ (100). Found, %: C 52.84; H 4.01; N 27.38. C9H8N4O2. Calculated, %: C 52.94; H 3.95; N 27.44.
1-Methyl-5-oxo-3-phenyl-4,5-dihydro-1Н-pyrazolo-[4,3-b]pyridine-6-carbonitrile (5g). Yield 0.61 g (49%, method I), 0.76 g (61%, method IІ), white powder, mp 285–288°C. IR spectrum, ν, cm–1: 3252 (N–H), 2230 (C≡N), 1671 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 4.07 (3H, s, NCH3); 7.38 (1H, t, J = 7.2, H-4 Ph); 7.47 (2H, t, J = 8.0, H-3,5 Ph); 7.85–8.08 (2H, m, H-2,6 Ph); 8.85 (1H, s, 7-CH); 12.15 (1H, s, NH). 13C NMR spectrum, δ, ppm: 37.0; 116.9; 126.8; 127.1; 128.5; 129.2; 129.9; 131.3; 136.9; 155.1; 159.9; 167.7. Mass spectrum, m/z (Irel, %): 251 [M+H]+ (100). Found, %: C 67.41; H 4.09; N 22.30. C14H10N4O. Calculated, %: C 67.19; H 4.03; N 22.39.
References
(a) Arora, N.; Billedeau, R. J.; Dewdney, N. J.; Gabriel, T.; Goldstein, D. M.; O'Yang, C.; Soth, M. US Patent 20050197340. (b) Arora, N.; Billedeau, R. J.; Dewdney, N. J.; Gabriel, T.; Goldstein, D. M.; O'Yang, C.; Soth, M.; Trejo-Martin, T. A. WO Patent 2007023105.
Duffy, B. C.; Liu, S.; Quinn, J. F.; Wang, R.; Jiang, M. X.; Martin, G. S.; Wagner, G. S.; Young, P. R. WO Patent 2015002754.
Blaquiere, N.; Burch, J.; Castanedo, G.; Feng, J. A.; Hu, B.; Staben, S.; Wu, G.; Yuen, P.-W. WO Patent 2015025025.
Betageri, R.; Cook, B. N.; Disalvo, D.; Harcken, C.; Kuzmich, D.; Liu, P.; Lord, J.; Mao, C.; Razavi, H. WO Patent 2012087782.
(a) Ibrahim, H. M.; Behbehani, H.; Makhseed, S.; Elnagdi, M. N. Molecules2011, 16, 3723. (b) Nag, S.; Nayak, M.; Batra, S. Adv. Synt. Catal.2009, 351, 2715.
(a) Turkman, N.; Shavrin, A.; Ivanov, R. A.; Rabinovich, B.; Volgin, A.; Gelovani, J. G.; Alauddin, M. M. Bioorg. Med. Chem.2011, 19, 5698. (b) del Mar Blanco, M.; Avendaño, C.; Cabezas, N.; Menéndez, J. C. Heterocycles1993, 36, 1387. (c) El-Sayed, O. A.; Aboul-Enein, H. Y. Arch. Pharm.2001, 334, 117. (d) El-Sayed, O. A.; Al-Bassam, B. A.; Hussein, M. E. Arch. Pharm.2002, 335, 403.
(a) Desos, P.; Lepagnol, J. M.; Morain, P.; Lestage, P.; Cordi, A. A. J. Med. Chem.1996, 39, 197. (b) Shi, J.; Xiao, Z.; Ihnat, M. A.; Kamat, C.; Pandit, B.; Hu, Z.; Li, P.-K. Bioorg. Med. Chem. Lett.2003, 13, 1187. (c) Tedesco, R.; Shaw, A. N.; Bambal, R.; Chai, D.; Concha, N. O.; Darcy, M. G.; Dhanak, D.; Fitch, D. M.; Gates, A.; Gerhardt, W. G.; Halegoua, D. L.; Han, C.; Hoffmann, G. A.; Johnston, V. K.; Kaura, A. C.; Liu, N.; Keenan, R. M.; Lin-Goerke, J.; Sarisky, R. T.; Wiggall, K. J.; Zimmerman, M. N.; Duffy, K. J. J. Med. Chem.2006, 49, 971. (d) Pathuri, G.; Li, Q.; Mohammed, A.; Gali, H.; Pento, J. T.; Rao, C. V. Bioorg. Med. Chem. Lett.2014, 24, 1380.
Yakovenko, G. G.; Lukianov, O. A.; Bol'but, A. V.; Vovk, M. V. Chem. Heterocycl. Compd.2019, 55, 379. [Khim. Geterotsikl. Soedin.2019, 55, 379.]
Watson, B. T.; Christiansen, G. E. Tetrahedron Lett.1998, 39, 9839.
Dagar, A.; Biswas, S.; Samanta, S. RSC Adv.2015, 5, 52497.
El Kazzouli, S.; Koubachi, J.; Berteina-Raboin, S.; Mouaddib, A.; Guillaumet, G. Tetrahedron Lett.2006, 47, 8575.
Yamanaka, T.; Ohki, H.; Ohgaki, M.; Okuda, S.; Toda, A.; Kawabata, K.; Inoue, S.; Misumi, K.; Itoh, K.; Satoh, K. WO Patent 2004101571.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2019, 55(12), 1211–1216
Rights and permissions
About this article

Cite this article
Yakovenko, G.G., Lukianov, O.А., Bol’but, A.V. et al. A convenient synthesis method of 5-oxopyrazolo[4,3-b]pyridine-6-carboxylic acids and their nitriles. Chem Heterocycl Comp 55, 1211–1216 (2019). https://doi.org/10.1007/s10593-019-02603-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-019-02603-5