
Abstract
Japanese eel (Anguilla japonica) is an important food source in East Asia whose population has dramatically declined since the 1970s. Despite past analysis with DNA sequencing, microsatellite and isozyme methods, management decisions remain hampered by contradictory findings. For example, it remains unresolved whether Japanese eels are a single panmictic population or whether they harbor significant substructure. Accurate assessment of population genetic substructure, both spatial and temporal, is essential for determining the relevant number of distinct management units appropriate for this species. In the present study, we assayed genetic variation genome-wide using Restriction Site Associated DNA Sequencing (RAD-seq) technology to analyze the population genetic structure of Japanese eels. For analysis of temporal isolation, five “cohort” samples were collected yearly from 2005 to 2009 in the Yangtze River Estuary. For analysis of spatial structure, five “arrival wave” samples were collected in China in 2009, and two arrival wave samples were collected in Japan in 2001. In each cohort of each arrival wave, five individuals were collected for a total of 55 eels sampled. In total, 214,210 loci were identified from these individuals, 106,652 of which satisfied quality checks and were retained for further analysis. There was relatively little population differentiation between arrival waves and cohorts collected either at different locations during the same year (Fst = 0.077) or at the same location collected over subsequent years (Fst = 0.082), and locations displayed no consistent isolation-by-distance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Temperate eels exhibit extraordinary life cycles, characterized by passive drifting of pelagic eggs and larvae over thousands of kilometers, followed by heroic homing migrations of adults back to natal waters. Japanese eels spawn at a single location in the Pacific Ocean (14–17°N, 142–143°E) between November and April each year (Tsukamoto 1992). Planktonic larvae follow the North Equatorial and the Kuroshio currents for 4 to 6 months, then metamorphose in pulses (known as “arrival waves”) into glass eels when they reach the coast of East Asia. This life history creates an opportunity for a pattern of genetic differentiation known as isolation-by-time (IBT) (Hendry and Day 2005). After living in their adult habitats for three or more years, they migrate 2000 to 3500 km back to their birthplaces to spawn. Other temperate eels such as the European eel (Anguilla anguilla) have a similar life history, with larvae who spawn in the Sargasso Sea, cross long distances following ocean currents to reach the Atlantic coast of Europe, and ultimately migrate about 5000–6000 km back to the birthplace to spawn and die (Kettle and Haines 2006).
Temperate eels are typical migratory fishes in that they show strong patchy habitat choice (Arai et al. 2004; Pujolar et al. 2011). Japanese eels inhabit various environments including fresh, brackish and coastal waters during their glass to yellow eel stages (Tsukamoto and Umezawa 1990). They can be divided into three different ecological types (Tsukamoto and Arai 2001; Tzeng et al. 2002): (1) sea eels (never enter freshwater), (2) estuary eels (stay in the estuary or frequently moved between freshwater and marine habitats), (3) river eels (typical catadromous fishes). European eel (A. anguilla), short-finned eel (A. australis) and New Zealand longfin eel (A. dieffenbachia) also have individuals that skip the freshwater stage and inhabit the sea or brackish water until their gonads begin to mature (Arai et al. 2004). Such spatial restriction to gene flow provides the opportunity for isolation-by-distance (IBD) (Rousset 1997). These observations beg the question, which pattern of genetic differentiation best describes the Japanese eels?
A limited number of eels contribute successfully to the next generation, as few individuals survive the long-distance catadromous migration and arrive to the spawning area near the Mariana Islands. Additionally, there is a brief window of 2–3 days before the new moon when eggs laid must be at the right maturity to survive. Differences in mating times within a reproductive season and patchy habitats may contribute to the restriction of gene flow among the different breeding batches or habitats (Hedgecock 1994; Hendry and Day 2005). Conversely, the fact that Japanese eels use a single spawning area and drift randomly in a highly mixed planktonic stage seems to suggest that panmixia might well be achieved despite the wide geographic range and temporal separation of generations. There are three conflicting hypotheses about population genetic structure of Japanese eels: (1) They are a single, panmictic population without any genetic heterogeneity at temporal and/or spatial scales. This hypothesis is supported by data from mtDNA sequences (Ishikawa et al. 2001). (2) They are a non-panmictic population with strong genetic substructure. This hypothesis is supported by the observation that Japanese eels collected from 7 locations throughout their range in northeastern Asia could be divided into a low-latitude group and a high-latitude group using eight microsatellite loci (Tseng et al. 2006). (3) They are a panmictic population, but harbor differences in local levels of genetic diversity over both temporal or spatial scales. Consistent with this hypothesis, studies using microsatellite loci found that varying levels of local genetic diversity were consistent with genetic drift in Japanese eels (Chan et al. 1997; Chang et al. 2007; Gong et al. 2014; Han et al. 2008, 2010a, b). Panmixia of European eel populations was initially accepted based on mtDNA sequence data (Avise et al. 1986), but was later challenged by significant population structuring found in more extensive microsatellite loci (Maes et al. 2006; Pujolar et al. 2013; Wirth and Bernatchez 2001). Subsequently, further evidence for panmixia of European eel was provided using genome-wide markers, based on RAD-sequencing data (Pujolar et al. 2014). The discrepancies among previous studies may be due to limited numbers of informative molecular markers, resulting in low power to detect genetic heterogeneity and population substructure (Maes et al. 2006; Pujolar et al. 2007, 2011; Pujolar et al. 2006).
Genome-wide SNP data obtained from Japanese eels sampled over time and space could resolve open questions about population structure. Restriction Site Associated DNA Sequencing (RAD-seq) is a cost effective method to assay variation in genome-wide SNPs (Miller et al. 2007, Baird et al. 2008), and has been shown to be highly informative for population structure analysis (Paschou et al. 2007). Here we apply RAD-seq to a collection of spatial and temporal samples of Japanese eels in an effort to identify the true population structure of these animals.
Materials and methods
Sampling
A total of 55 Japanese eels (11 cohorts of 5 individuals each) were collected for RAD sequencing from Japan and China. The Japanese cohorts included two arrival waves in 2001 collected at different locations, and the Chinese cohorts included five arrival waves in 2009 collected at different locations. This sampling effort covers most of the Japanese eel range, spanning from Japan to Southern China, and is either broader or similar in scope to previous genetic studies of Japanese eel (Ishikawa et al. 2001; Tseng et al. 2006; Gong et al. 2014; Han et al. 2008, 2010a, b). While our sampling efforts do not include samples from the Korean peninsula as several previous studies (Tseng et al. 2006; Han et al. 2010a), we collect samples from as far south as Guangdong Province, China. In addition to geographic samples, intra-annual samples were drawn from 6 cohorts (eels from a breeding season), including 1 from Japan in 2001 and 5 from Yangtze River estuary during 2005 to 2009 between January and March (Table 1). All samples were glass eels except for the 2007 and 2008 samples from the Yangtze River Estuary, which were ages 5 and 4 (silver eels), respectively. These were caught in the Jingjiang River, China (31°55′N, 120°12′E) in October 2012 as they were migrating back to spawn. We collected five giant mottled eels (Anguilla marmorata) from Hainan Province, China in July 2014 to serve as the outgroup (Table 1; Fig. 1). Living glass eels and the caudal fins of the silver eels were washed several times in distilled water and preserved in 95% ethanol. Genomic DNA was extracted using a standard phenol–chloroform protocol (Sambrook and Russell 2006). The quality and quantity of DNA from each sample were measured with a 1% agarose gel and a TBS-380 Mini-Fluorometer using PicoGreen (Fig. 2).

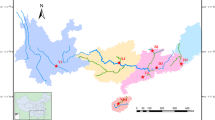
Sampling locations and years for the samples of Anguilla japonica used in this study. Each dot represents one sampling location. The horizontal row of five blue dots represents five cohorts from the Yangtze River during 2005–2009. The five vertical, blue dots of varied hues represent five arrival waves from the coastal area of China in 2009. The light blue dot represents the Giant mottled eel from Hainan, China in 2014. a The geographic location of each cohort. b The years of collection for each cohort. (Color figure online)

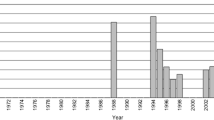
Variant site (SNP) counts of every sampling group using Stacks depths of 3, 6, 10. The light, middle, and dark grey shading represent depths 3, 6, and 10, respectively
RAD-sequencing
The RAD libraries for each sample were prepared following protocols from (Baird et al. 2008; Miller et al. 2007; Pujolar et al. 2013). Genomic DNA from each individual was digested with the restriction enzyme KpnI. Barcoded adapters were ligated onto the fragments in order to multiplex samples. All samples were pooled and fragmented to 300–400 bp with 0.3 MPa pressurized nitrogen. A second adaptor was ligated to the sheared fragments and products were PCR amplified prior to sequencing. Samples were multiplexed into two separate libraries and sequenced on an Illumina HiSeq2000. Both libraries were sequenced as 100 bp single-end reads, and one library was resequenced as a paired-end library with 125 bp reads. Forward reads from the paired-end run were trimmed to 100 bp using fastx_trimmer from FASTX Toolkit 0.0.13. Raw sequencing reads have been deposited at Dryad (accession numbers available upon acceptance of publication). Reads were demultiplexed and quality filtered using the ‘process_radtags’ script from Stacks 1.48.
Calling and filtering SNPs
Stacks-1.48 was used to call and analyze SNPs from the RAD-seq data (Catchen et al. 2013).
Preparing data for Stacks
The Stacks script process_radtags was run to QC the reads and demultiplex the samples. Any read with an uncalled base and low quality scores was discarded with filtered-data options –c –q--inline_null (-c removes reads with uncalled bases, -q discards reads with low quality scores, --inline_null indicates that barcodes were included in the read rather than separate file). The Japanese eel reference genome (GenBank: AVPY00000000.1) (Henkel et al. 2012) was downloaded, and the mitochondrial genome was separated from the whole genome source file using BBmap-35.66. Retained reads were mapped to the nuclear genome using Bowtie-1.1.2 (Langmead et al. 2009). No more than two mismatches per read were accepted. Those reads that failed to map to the nuclear genome were submitted for mapping to the mitochondrial genome. An average of 1,130,254 reads per individual mapped to the nuclear reference genome, but for sample JJ-107 from Yangtze-2008, this count was only 11,012, leading us to drop this sample. An average of 854 reads from each sample were mapped to the mitochondrial genome.
We tested several stack depths in order to optimize our analysis of RAD-seq data, as recommended (Catchen et al. 2013). Stack depths of 3, 6, 10 were set to call SNPs. The following analyses were run on each depth:
Running Stacks to call and correct SNPs
i) Reference-aligned reads were pre-processed using the ref_map.pl pipeline in Stacks-1.48; ii) Then the rxstacks program was run to make corrections to genotype and haplotype calls in individual samples based on data accumulated from a population-wide examination. The rxstacks program options for SNP quality were set as follows: --lnl_lim -10, --conf_lim 0.25, --prune_haplo, --bound_high 0.1, --verbose, --conf_filter, --prune_haplo; iii) cstacks and sstacks were rerun to reassemble the catalog. iv) The populations program was run for each depth 3, 6, and 10 (-m) with parameters -p 1, -r 0.6, --write_single_snp, --fstats, --fst_correction ‘p_value’. Samples were assigned to 12 different populations, representing the 11 Japanese eel cohorts and the 1 outgroup cohort. The samples with low calls and loci with high depth were filtered out, and the populations were then re-run. We then filtered out any locus having a depth more than two SD higher than the mean coverage. Mean coverage across loci was 11.35 for—m3, 14.59 for—6, and 19.4 for—m10. Only 1 SNP was identified in the mitochondrial genome and it was discarded. In the end there were 106,652 biallelic SNPs carried forward for subsequent analysis with an average depth of 7.76 reads per locus across individuals.
The population genetic structure analysis
The genetic diversity of each population was calculated using the genome-wide SNP data, including nucleotide diversity (π), Fst, observed (Ho) and expected (He) heterozygosities in Stacks (Fig. 3). Additionally, we divided the Japanese eel samples into four geographically relevant “meta-populations” to increase sample sizes within each population and calculated diversity statistics and Fst using the population module of Stacks (“South China Sea” includes Guangdong2009/09; “East China Sea” includes Fujian2009/09, Yangtze2005/05, Yangtze2006/06, Yangtze2007/07, Yangtze2008/08, Yangzte2009/09; “Yellow Sea” includes Jiangsu2009/09; and “Pacific Ocean” includes Chibaken2001/01 and Kagawa2001/01). The genetic diversity differences among samples were tested by one-way ANOVA followed by Tukey Honest Significant Differences using R (v. 3.4.4). IBD (isolation by distance) and isolation by cohorts were tested using Mantel tests (Mantel 1967) by correlating the linearized genetic distance Fst/ (1-Fst) versus geographic distance and sampling years using mantel.test from the R package ape v5.1. Principal Component Analysis (PCA) was run using Plink 1.09_beta3.30 (Purcell et al. 2007).

Genetic diversity of Japanese eels among 11 sampling groups. Ho observed heterozygosity; He expected heterozygosity; π nucleotide diversity. These values were all calculated at only the variable sites in each population. The different color bars represent different sampling groups. (Color figure online)
Computer code for execution of all analysis in this project can be found on Github at https://github.com/erdavenport/japaneseEel.
Results
SNP calling
A total of 160 million raw reads were generated for the 60 samples, 125 million reads (78%) were retained and 35 million reads (22%) were discarded after removing ambiguous barcodes, low quality, and excess ambiguous RAD-tags. Of those reads, 68 million reads (54%) were aligned to the nuclear genome, 42 million (34%) failed to align, and 14 million reads (12%) clustered to a depth less than 3. In total, 57% of raw reads were discarded (Table 2). Sequence raw data, sample information, and vcf files were deposited into the Dryad Archive (http://datadryad.org/).
Optimal Stacks parameters vary by experiment, and we wanted to identify the set of parameters that would be the most useful to our analysis. One of the factors that matters is the number of reads used to call a stack (stack depth) and the analysis was run at depths of 3, 6, and 10. The number of SNPs in the final catalog at stack depths 3, 6, 10 were 106,652, 93,788, and 75,193, respectively. Results of Fst analysis at depths of 3, 6, 10 were consistent with each other, and because we retain the most SNPs at a minimum stack depth of 3, all subsequent analysis of population structure of the Japanese eel used SNPs called with a minimum stack depth of 3.
There were 805,573 raw SNPs called from the 68 million retained reads. 795,991 SNPs were retained after running the corrected module rxstacks. 686,486 SNPs were filtered out by the Stacks module populations with the options p = 1, r = 0.6, and –write_single_snp. Only 2,853 SNPs were discarded because they had more than two SD higher than the mean coverage, and in the end 106,652 SNPs were used to analyze the population genetic structure of Japanese eel (Table 2).
Population genetic structure analysis
Genetic diversity
Japanese eels inhabit a wide variety of environments, so one might expect to see some degree of genetic differentiation across geographic regions. The observed heterozygosity (Ho) within each geographic sample ranged from 0.0004 ± 0.0001 to 0.0007 ± 0.0003, with a mean of 0.0006 ± 0.00008. The highest diversity was observed in Yangtze River estuary in 2008, and the lowest diversity was found in Kagawa in 2001. We note that the Kagawa had the lowest coverage of all of the population groups, which could explain why the lowest diversity was observed in these individuals. The same patterns both occurred in expected heterozygosity (He) (which ranged from 0.0005 ± 0.0001 to 0.0008 ± 0.0003, with a mean of 0.0007 ± 0.0001) and nucleotide diversity (π) (which ranged from 0.0006 ± 0.0002 to 0.0008 ± 0.0003 with a mean of 0.0008 ± 0.0001).
Significant differences (P < 0.05) were observed in contrasts of within-sample genetic diversity measures (Ho, He, and π) across many of the pairs of samples (ANOVA in R). For ease of presentation, only the comparisons that were not significant (P > 0.01) are presented in Supplementary Table 1.
Fst is a standardized variance, representing the portion of the total genetic variance that is due to among-subpopulation differences (Weir 1996). The overall mean Fst value across all pairs of population samples was 0.079. The pairwise average Fst was 0.077 across the 5 arrival waves in China 2009, and across the 5 cohorts from Yangtze River estuary during 2005 ~ 2009, Fst was 0.082 (Supplementary Table 2). Additionally, we divided the small cohort samples into four “meta-populations” (South China Sea, East China Sea, Yellow Sea, and Pacific Ocean, see Materials and Methods) to increase sample size within each population. Fst between each of these larger groups remained low (between 0.015 East China Sea – Pacific Ocean and 0.076 South China Sea – Yellow Sea), further supporting the conclusion of a homogeneous small departure from panmixia.
Isolation-by-distance (IBD) or by spatial scale and isolation by temporal scale
Japanese eels inhabit various environments which opens the opportunity for genetic divergence. Even though they breed in a common breeding area in the Pacific, this does not necessarily guarantee panmixia within the breeding area. Moreover, the reproductive time within a reproductive season varies, and few adults successfully contribute to recruitment, as few individuals make it back to the spawning ground. Despite this opportunity for genetic differentiation, the data show no significant correlation between linearized genetic distance Fst/(1–Fst) and temporal size, measured as number of years between cohorts sampled from Yangtze River estuary during 2005–2009 (Mantel test, P = 0.211) (Fig. 4a). The result was the same when the temporal dimension was extended to 2001, and the samples from Japan were included (figure not shown). The IBD test for association between genetic distance and geographic distances was not found to be significant (P = 0.315) (Fig. 4b). So it appears that there is random mating with respect to geography, but Fst is persistently greater than zero because of sweepstakes reproduction perturbing allele frequencies at each site (Fig. 5).

Mantel test of the correlation of pairwise linearized genetic distance (Fst/(1-Fst)) vs. sampling years (a) and the spherical earth distance (b) in kilometers. The 2006,2007,2008 and 2009 at the horizontal axis represent the 1,2,3 and 4 years distance between sampling groups separately

The UPGMA tree based on the genetic distance of the 106,652 RAD-seq retained loci the same color circles represent the same sampling groups. We used the giant mottled eel (Hainan province-8-2014) as an outgroup. (Color figure online)
Phylogenetic tree. The evolutionary relationships among all groups of Japanese eel can be represented by a phylogenetic tree
The 55 Japanese eel and the five giant mottled eel data were used to calculated a ‘pair-wise’ matrix of Fst values, which was then used to construct a UPGMA (Unweighted Pair Group Method with Arithmetic Mean) phylogenetic tree. Not surprisingly, the giant eels formed a clear outgroup lineage, and the Japanese eels fell into one clade with nearly equidistant “tips” and with no clustering based either on time or geography (Fig. 3).
Discussion
A single, panmictic population
The population structure and mating patterns of Japanese eels have been interrogated using a variety of genetic markers and sample sources, however in the past there has been no consensus as to whether the species is truly panmictic (Chang et al. 2007; Gong et al. 2014; Han et al. 2008, 2010a; Ishikawa et al. 2001; Sang et al. 1994; Tseng et al. 2006). In this study, genome-wide SNPs were interrogated across samples collected from different regions and years to determine genetic diversity, differentiation, and structure of the Japanese eel. Our results demonstrate that Japanese eels exist as a panmictic population. First, the pairwise Fst comparisons between sampling cohorts were exceedingly low, all falling in a small range around 0.075. s, the IBD Mantel tests showed no significant heterogeneity across samples (P > 0.05), either across years or across geography, despite the samples’ broad geographical distribution from northern Japan to southern Taiwan. There has been much discussion about the differentiation in both time and space of the Japanese eel populations (Chang et al. 2007; Gong et al. 2014; Han et al. 2008, 2010b; Ishikawa et al. 2001; Sang et al. 1994; Tseng et al. 2008). Han et al. (2010a) reported that there was a slight year-to-year genetic variation among 21 yearly cohorts (P = 0.027), but this temporal variation could be explained by random genetic drift across generations, and is not reflective of a departure from panmixia. Tseng et al. (2006) suggested that there were separate high-latitude and low-latitude groups, but this conclusion has been questioned because Tseng et al. used loci deviating from Hardy–Weinberg equilibrium (Han et al. 2010b). In short, all the results presented here support the hypothesis that the Japanese eel exists as a single panmictic population, and the small, consistent level of inter-population heterogeneity is probably driven by sweepstakes reproduction in each local area (see below).
Genetic heterogeneity
Many studies suggested that a panmictic population might nevertheless possess geographic differences in the local amount of genetic variation. Fluctuating genetic patchniess (Hellberg et al. 2002) in species with pelagic larvae may be accounted for by variation in settlement patterns (Edmands et al. 1996; Hedgecock 1994; Johnson and Black 1982; Moberg and Burton 2000) which at any one time may appear as differences in local levels of genetic diversity. The larvae of the Angullia genus are passively spread by ocean currents and randomly choose their environments, which perhaps could lead to local differences in genetic diversity. But genetic heterogeneity over a 21 year time scale in Japanese glass eels was only marginally significant using eight microsatellite loci, and the overall level of variation was consistent with random genetic drift (Han et al. 2010a). Similar levels of patchiness in genetic diversity were also observed in the European eel both among cohorts and among samples within cohorts using 10 allozyme and 6 microsatellite loci (Pujolar et al. 2006), it was especially strong in maturing adults of the European eel using 22 EST-linked microsatellite loci (Pujolar et al. 2011).
In this study, only a few pairwise comparisons among samples were found to display the same level of genetic diversity (P > 0.01) (Supplementary Table 1). Three mature silver eels were outliers on the PCA plot (Fig. 6), consistent with some degree of temporal genetic differentiation between mature adult eels and glass eels. As with many other marine organisms, Japanese eels possess an extensive geographical distribution, long lifespan, late maturation, and a small number of survivors that reach their spawning sites to successfully produce many offspring. This kind of reproductive pattern has been described as “sweepstakes reproductive success (SRS)” (Ragauskas and Butkauskas 2013). Under the SRS hypothesis, the random variation in parental contribution to the next generation leads to unpatterned variation in genetic composition of recruits (genetic patchiness) (Pujolar et al. 2011). The genetic heterogeneity of Japanese eel was likely result from the SRS (Hedgecock 1994), but this genetic heterogeneity is re-set each generation by panmixia in the breeding site.

The relationship among all individuals of Japanese eel based on the first and second principal coordinates. Individuals colored by location of sampling. 3 individuals are outliers. (Color figure online)
One concern with the present study is the relatively small sample sizes and depth of sequencing. Our cohort sizes were limited by availability of biological samples spanning historic and geographic distances (n = 5 per cohort). However, population differentiation values such as Fst can be accurately calculated for small sample sizes, given that the number of biallelic markers examined is high (n > 1000) (Willing et al. 2012). While the per population sample size is low in each of the waves and cohorts in this study, the number of loci examined is high (> 100,000 loci), providing accurate estimates of population differentiation. Furthermore, it is expected that small sample sizes would lead to an overestimation of genetic differentiation, not the panmixia reported here. Our simulations confirm this gain in accuracy from sampling of larger numbers of loci (see Supplementary Table 3). Additionally, although our sequencing depth is modest (7x at the lowest minimum stack depth), we report similar within-species Fst values as previous studies performed using nuclear microsatellite markers in Japanese eel and in the European eel (Tseng et al. 2006; Baltazar-Soares et al. 2014) .
In short, the extensive sampling and deep RAD-seq SNP ascertainment in our study reveals a pattern of geographic differentiation consistent with panmixia in the breeding population, followed by dispersal and random perturbations of allele frequencies that are remarkably consistent over both spatial and temporal scales. This inconsistency of allele frequency changes over time and space lead us to conclude that there is a lack of evidence supporting local adaptation in the Japanese eel. This makes the task of management of this commercially important species far more tractable.
References
Arai T, Kotake A, Lokman PM, Miller MJ, Tsukamoto K (2004) Evidence of different habitat use by New Zealand freshwater eels Anguilla australis and A. dieffenbachii, as revealed by otolith microchemistry. Mar Ecol Prog Ser 266:213–225. https://doi.org/10.3354/meps266213
Avise JC, Helfman GS, Saunders NC, Hales LS (1986) Mitochondrial DNA differentiation in North Atlantic eels: population genetic consequences of an unusual life history pattern. Proc Natl Acad Sci USA 83:4350–4354. https://doi.org/10.1073/pnas.83.12.4350
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3:1–7. https://doi.org/10.1371/journal.pone.0003376
Baltazar-Soares M, Biastoch A, Harrod C, Hanel R, Marohn L, Prigge E, Evans D, Bodles K, Behrens E, Böning CW, Eizaguirre C (2014) Recruitment collapse and population structure of the european eel shaped by local ocean current dynamics. Curr Biol 24:104–108. https://doi.org/10.1016/j.cub.2013.11.031
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140. https://doi.org/10.1111/mec.12354
Chan I, Chan D, Lee S, Tsukamoto K (1997) Genetic variability of the Japanese eel Anguilla japonica (Temminck & Schlegel) related to latitude. Ecol Freshw Fish 6:45–49. https://doi.org/10.1111/j.1600-0633.1997.tb00141.x
Chang K, Han Y, Tzeng W (2007) Population genetic structure among intra-annual arrival waves of the Japanese Eel. Zool Stud 46:583–590
Edmands S, Moberg PE, Burton RS (1996) Allozyme and mitochondrial DNA evidence of population subdivision in the purple sea urchin Strongylocentrotus purpuratus. Mar Biol 126:443–450. https://doi.org/10.1007/BF00354626
Gong X, Ren S, Cui Z, Yue L (2014) Genetic evidence for panmixia of Japanese eel (Anguilla japonica) populations in China. Genet Mol Res 13:768–781. https://doi.org/10.4238/2014.January.31.3
Han Y, Sun Y, Liao Y, Tzeng WN (2008) Temporal analysis of population genetic composition in the overexploited Japanese eel Anguilla japonica. Mar Biol 155:613–621. https://doi.org/10.1007/s00227-008-1057-1
Han Y, Hung C, Liao Y, Tzeng W (2010a) Population genetic structure of the Japanese eel Anguilla japonica: panmixia at spatial and temporal scales. Mar Ecol Prog Ser 401:221–232. https://doi.org/10.3354/meps08422
Han Y, Iizuka Y, Tzeng W (2010b) Does variable habitat usage by the Japanese eel lead to population genetic differentiation? Zool Stud 49:392–397
Hedgecock D (1994) Does variance in reproductive success limit effective population sizes of marine organisms? Genet Evol Aquat Org 122–134
Hellberg ME, Burton RS, Neigel JE, Palumbi SR (2002) Genetic assesment of connectivity among marine populations. Bull Mar Sci 70:273–290
Helyar SJ, Hemmer-Hansen J, Bekkevold D, Taylor MI, Ogden R, Limborg MT, Cariani A, Maes GE, Diopere E, Carvalho GR, NielEE (2011) Application of SNPs for population genetics of nonmodel organisms: New opportunities and challenges. Mol Ecol Resour 11:123–136. https://doi.org/10.1111/j.1755-0998.2010.02943.x
Hendry AP, Day T (2005) Population structure attributable to reproductive time: Isolation by time and adaptation by time. Mol Ecol 14:901–916. https://doi.org/10.1111/j.1365-294X.2005.02480.x
Henkel CV, Dirks RP, de Wijze DL, Minegishi Y, Aoyama J, Turner B, Knudsen BJ, Bundgaard M, Hvam K, Boetzer M, Pirovano W, Weltzien F, Dufour S, Tsukamoto K, Spaink HP, van den Thillart GE (2012) First draft genome sequence of the Japanese eel, Anguilla japonica. Gene 511:195–201. https://doi.org/10.1016/j.gene.2012.09.064
Ishikawa S, Aoyama J, Tsukamoto K, Nishida M (2001) Population structure of the Japanese eel Anguilla japonica as examined by mitochondrial DNA sequencing. Fish Sci 67:246–253
Johnson MS, Black R (1982) Chaotic genetic patchiness in an intertidal limpet, Siphonaria sp. Mar Biol 70:157–164. https://doi.org/10.1007/BF00397680
Kettle AJ, Haines K (2006) How does the European eel (Anguilla anguilla) retain its population structure during its larval migration across the North Atlantic Ocean? Can J Fish Aquat Sci 63:90–106. https://doi.org/10.1139/f05-198
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. https://doi.org/10.1186/gb-2009-10-3-r25
Maes GE, Pujolar JM, Hellemans B, Volckaert FAM (2006) Evidence for isolation by time in the European eel (Anguilla anguilla L.). Mol Ecol 15:2095–2107. https://doi.org/10.1111/j.1365-294X.2006.02925.x
Mantel N (1967) The detection of disease clutersing and a generalized regression approach. Cancer Res 27:209–220
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA (2007) Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res 17:240–248. https://doi.org/10.1101/gr.5681207
Moberg PE, Burton RS (2000) Genetic heterogeneity among adult and recruit red sea urchins, Strongylocentrotus franciscanus. Mar Biol 136:773–784. https://doi.org/10.1007/s002270000281
Paschou P, Ziv E, Burchard EG, Burchard FG, Choudhry S, Rodriguez-Cintron W, Mahoney MW, Drineas P (2007) PCA-correlated SNPs for structure identification in worldwide human populations. PLoS Genet 3:1672–1686. https://doi.org/10.1371/journal.pgen.0030160
Pujolar JM, Maes GE, Volckaert FAM (2007) Genetic and morphometric heterogeneity among recruits of the European eel, Anguilla anguilla. Mar Ecol Prog Ser 81:297–308. https://doi.org/10.3354/meps307209
Pujolar J, Daniele B, Andrello M, Capoccioni F, Ciccotti E, Leo GAD, Zane L (2011) Genetic patchiness in European eel adults evidenced by molecular genetics and population dynamics modelling. Mol Biol Evol 58:198–206. https://doi.org/10.1016/j.ympev.2010.11.019
Pujolar JM, Jacobsen MW, Frydenberg J, Als TD, Larsen PE, Maes GE, Zane L, Jian JB, Cheng L, Hansen MM (2013) A resource of genome-wide single-nucleotide polymorphisms generated by RAD tag sequencing in the critically endangered European eel. Mol Ecol Resour 13:706–714. https://doi.org/10.1111/1755-0998.12117
Pujolar JM, Jacobsen MW, Als TD, Frydenberg J, Magnussen E, Jónsson B, Jiang X, Cheng L, Bekkevold D, Maes GE, Bernatchez L, Hansen MM (2014) Assessing patterns of hybridization between North Atlantic eels using diagnostic single-nucleotide polymorphisms. Heredity 112:627–637. https://doi.org/10.1038/hdy.2013.145
Pujolar1 JM, Maes1 GE, Volckaert1 FAM (2006) Genetic patchiness among recruits in the European eel Anguilla anguilla. Mar Ecol Prog Ser 307:209–217
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575. https://doi.org/10.1086/519795
Ragauskas A, Butkauskas D (2013) The formation of the population genetic structure of the European eel Anguilla anguilla (L.): a short review. Ekologija 59:143–154
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Sambrook J, Russell DW (2006) Purification of nucleic acids by extraction with phenol:chloroform. CSH Protoc. https://doi.org/10.1101/pdb.prot4455
Sang T, Chang H, Chen C, Hui C (1994) Population structure of the Japanese eel, Anguilla japonica. Mol Biol Evol 11:250–260
Tseng M, Tzeng W, Lee S (2006) Population genetic structure of the Japanese eel Anguilla japonica in the northwest Pacific Ocean: evidence of non-panmictic populations. Mar Ecol Prog Ser 308:221–230. https://doi.org/10.3354/meps308221
Tseng M, Tzeng W, Lee S (2008) Genetic differentiation of the Japanese Eel. Am Fish Soc Symp 58:59–69
Tsukamoto K (1992) Discovery of the spawning area for Japanese eel. Nature 356:789–791. https://doi.org/10.1038/356789a0
Tsukamoto K, Arai T (2001) Facultative catadromy of the eel Anguilla japonica between freshwater and seawater habitats. Mar Ecol Prog Ser 220:265–276. https://doi.org/10.3354/meps220265
Tsukamoto K, Umezawa A (1990) Early life history and oceanic migration of the eel Anguilla japonica. La mer 188–198
Tzeng W, Shiao J, Iizuka Y (2002) Use of otolith Sr:Ca ratios to study the riverine migratory behaviors of Japanese eel Anguilla japonica. Mar Ecol Prog Ser 245:213–221. https://doi.org/10.3354/meps245213
Weir BS (1996) Genetic data analysis II: methods for discrete population genetic data. Sinauer Assoc Sunderl MA 150–156. https://doi.org/10.1136/jmg.29.3.216
Willing EM, Dreyer C, van Oosterhout C (2012) Estimates of genetic differentiation measured by Fst do not necessarily require large sample sizes when using many SNP markers. PLoS ONE 7:1–7. https://doi.org/10.1371/journal.pone.0042649
Wirth T, Bernatchez L (2001) Genetic evidence against panmixia in the European eel. Nature 409:1037–1040. https://doi.org/10.1038/35059079
Acknowledgements
We sincerely thank Dr. Minhui Wang for assistance with data analysis. Research supported by the Cornell China Faculty Development Program,the National Natural Science Foundation of China (31201995), the China-ASEAN Maritime Cooperation Fund from Shanghai University, the Shanghai Municipal Agricultural Commission (No. 2013 2–2), the Shanghai Municipal Science and Technology Commission of Chongming (Grant No. 13231203504) and the Open Foundation of Engineering Research Centre of Modern Industrial Technology for Eels (No. RE201501), Ministry of Education. ERD is supported by NIH F32 DK109595.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gong, X., Davenport, E.R., Wang, D. et al. Lack of spatial and temporal genetic structure of Japanese eel (Anguilla japonica) populations. Conserv Genet 20, 467–475 (2019). https://doi.org/10.1007/s10592-019-01146-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-019-01146-8