
Abstract
The mountain yellow-legged species complex (Rana sierrae and Rana muscosa) has declined precipitously in distribution and abundance during the last century. The two primary threats are chytrid epidemic-associated population collapses and predation from the introduction of non-native trout. Widespread declines have occurred throughout the ranges of these species, including populations of R. sierrae in Yosemite National Park. A clear picture of genetic structure of remaining Yosemite R. sierrae populations is critical to short-term management and conservation. We conducted a population genetics study that included samples from 23 geographic sites distributed throughout the range of R. sierrae in Yosemite NP. We used minimally-invasive swab samples to collect genetic data from mitochondrial and nuclear DNA via sequencing (43 transcriptome-derived markers) and analyzed the distribution of genetic variation in a geographic context. Our mtDNA analysis partially confirmed previous results suggesting that two haplotype groups occur in Yosemite: one haplotype group contained high bootstrap support for monophyly while the other did not. However, increased geographic sampling demonstrated that the two haplotypes are not completely geographically partitioned into the two main drainages (Merced drainage and Tuolumne drainage) as previously postulated. Our nuclear DNA analysis revealed a general pattern of genetic isolation by distance, where genetic differentiation was correlated with geographic distance between sites. In addition, our analyses suggested that three clusters of genetically cohesive sites occur in the study area. Understanding population genetic patterns of variability will inform management strategies such as translocations, reintroductions, and monitoring for this endangered frog. Lastly, our next generation sequencing enabled approach allowed us to obtain multi-locus data from minimally-invasive swab samples. Thus researchers can now leverage extensive archives of swab samples (initially collected for pathogen testing) to study host genetics in previously surveyed amphibian populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amphibians are in decline worldwide, with more than 30% of species categorized as globally threatened with extinction (Stuart et al. 2004). Reversing such declines is challenging and will often require detailed information on the species of interest, including historical and current distribution, natural history, threats, and population characteristics affecting long-term persistence (Semlitsch 2002). In addition, an understanding of species’ phylogeography is essential if historical genetic variation is to be preserved or restored (e.g., Shaffer et al. 2004; Lind et al. 2011). Baseline phylogeographic data also serve as a foundation for developing recovery actions such as reintroductions and captive rearing designed to reestablish extirpated populations (McCartney-Melstad and Shaffer 2015).
Mountain yellow-legged frogs (Rana muscosa and Rana sierrae) are emblematic of the global amphibian decline crisis. Historically, they were among the most abundant vertebrate species in the high elevation Sierra Nevada (Grinnell and Storer 1924). Rana muscosa/sierrae currently are found at only 7% of their historical localities, despite the remote and undeveloped characteristics of the associated landscape (e.g. national parks, wilderness areas) (Vredenburg et al. 2007). As a consequence of this precipitous decline, both species are now listed as “endangered” under the federal Endangered Species Act, while the California Endangered Species Act lists R. sierrae as “threatened” and R. muscosa in the Sierra Nevada is listed as “endangered”.
The two primary drivers of population declines for mountain yellow-legged frogs are the introduction of non-native predatory trout in lakes and streams and the arrival of the infectious disease chytridiomycosis caused by the amphibian chytrid fungus (Batrachochytrium dendrobatidis; “Bd”). Starting in the mid-1800s trout were introduced into thousands of naturally fishless lakes in the Sierra Nevada, many of which contained native R. muscosa/sierrae populations. Co-occurrence of introduced trout and frog populations typically resulted in subsequent frog population decline and extirpation due to predation by the non-native trout (Knapp and Matthews 2000; Knapp 2005). Although fish stocking ceased in National Parks by the early 1990s, many introduced fish populations persist in aquatic habitats that would otherwise be suitable habitat for R. muscosa/sierrae (e.g., lakes, low-gradient streams). Invasive pathogens are also a dramatic threat, and the effect of Bd on mountain yellow-legged frogs provides one of the most striking examples of chytrid-associated amphibian population declines in North America. During the past several decades Bd has spread across the Sierra Nevada and caused the decline or extirpation of hundreds of mountain yellow-legged frog populations (Vredenburg et al. 2010). In response to these population declines, academic scientists and natural resource managers from state and federal agencies have led management efforts including population monitoring, non-native fish removal, Bd surveys, captive breeding, and bacterial augmentation experiments to reduce Bd infection intensities (e.g., Knapp and Matthews 2000; Vredenburg et al. 2010).
A thorough understanding of the genetic structure of remaining R. muscosa/sierrae populations is critical to management and conservation of these endangered species. Information on population structure will be important to determine if subpopulations are demographically independent and therefore warrant consideration as separate management units. Genetic data can also be used to identify suitable donor populations to re-establish frogs in areas from which they were previously extirpated and provide supplemental frogs to areas with low population sizes (Knapp et al. 2011).
Previous work by Vredenburg et al. (2007) analyzed the phylogeography of the R. muscosa/sierrae species complex using a mitochondrial DNA (mtDNA) marker. Their analysis found three mtDNA clades of each species (five clades in the Sierra Nevada, one clade in southern California) in a pattern concordant with geographic distribution of the populations. Although Vredenburg et al. (2007) provided important insights into the genetic structure of this species complex, an updated analysis of population structure in the Sierra Nevada—and Yosemite National Park in particular—is needed. Vredenburg et al. (2007) relied on a single mtDNA marker, and inferences based on mtDNA can be discordant with patterns in the nuclear genome due to different inheritance patterns (mtDNA is maternally inherited) (Toews and Brelsford 2012). Additionally, Vredenburg et al. (2007) sought to characterize genetic variation at broad geographic scale and included a relatively small number of samples for focal regions in the Sierra Nevada. For example, R. sierrae in Yosemite National Park was represented by only seven samples. Thus additional genomic and geographic sampling is needed to understand fine scale patterns of genetic variation.
We sought to better resolve the genetic structure of R. sierrae populations in Yosemite National Park using mitochondrial data and a newly developed multi-locus nuclear assay. Based on the R. muscosa/sierrae transcriptome that we sequenced previously (Rosenblum et al. 2012), we developed a new high throughput assay to target dozens of nuclear markers. Importantly, this new assay works well with low concentration samples, allowing us to use minimally-invasive skin swab samples instead of toeclips. To understand the genetic structure of remaining R. sierrae populations, we generated genetic data for 192 frogs from 23 sites spread across Yosemite. Vredenburg et al. (2007) found that Yosemite contains two mtDNA clades, which are separated into the two drainages in Yosemite: Tuolumne River drainage in the northern region and Merced River drainage in the southern region. This spatial pattern of genetic variation in mtDNA suggested little or no connectivity between drainages. We tested this a priori two-population hypothesis with our dataset that contains larger sample sizes and multi-locus genetic data. We also used genetic clustering methods that are agnostic to sampling location in order to let the genetic data reveal the structure of populations in Yosemite. We then used spatially-explicit methods to analyze the spatial distribution of genetic variation, assess connectivity between drainages, and test for isolation by distance among sampling locations.
Methods
Sampling scheme
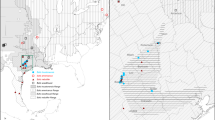
We used archived genetic samples obtained from skin swabs that were previously collected for monitoring Bd infection in R. sierrae populations in Yosemite National Park (results reported in Briggs et al. 2010; Vredenburg et al. 2010). We supplemented the swab archive by collecting additional swab samples from under-sampled geographic areas of Yosemite. For mitochondrial DNA sequencing, we included 106 individuals from the 23 sampling sites. For nuclear DNA sequencing, we included 192 individuals from 23 R. sierrae populations with 3–15 samples per location (Fig. 1, Supplementary Table 1). Figure 1 shows the sample grouping scheme, where the sampling sites are labeled with numbers that are roughly ordered in a clockwise pattern on the landscape. Sites represent either single sample locations (e.g., a single lake) or groups of sample locations within relatively close proximity (e.g., ponds and/or streams within a watershed basin). Euclidean distances between sites ranged from 2.2 to 61.0 km with an average of 29.7 ± 15.5 km (mean ± standard deviation). We note that genetic clustering data analyses (described below) were agnostic to sampling site assignment and therefore avoided biased grouping of samples.

Yosemite National Park study area, showing the sample locations (circles). Site labels (black squares) represent either single sample locations or contain groupings of nearby sample locations as indicated by connecting lines to sampling locations. Shaded label boxes indicate drainage location: grey Tuolumne, black Merced. The locations of the Tuolumne River and the Merced River are specified with arrows
Nucleic acid preparation and purification
Swabs were stroked 30 times on the dorsal and ventral surface of the sampled frogs, and nucleic acids were extracted from swabs with the Prepman Ultra reagent (Hyatt et al. 2007). To further purify DNA from the swab extracts, we used an isopropanol precipitation protocol as described in the Prepman Ultra manual appendix. Briefly, we centrifuged the extract at 16,000×g for 2 min to pellet debris, and moved the supernatant to a fresh low-adhesion tube. We added low TE buffer to a final volume of 447.5 uL (10 mM Tris–HCl, 0.1 mM EDTA; note that EDTA concentration is lower than typical in order to avoid PCR inhibition). Then we added 2.5 uL of 20 ug/uL glycogen and 50 uL of 3 M NaOAc, pH 5.2, gently mixed, then added 500 uL of isopropanol, and gently mixed again. After 20 min of room temperature incubation, we centrifuged the tubes at 13,000×g for 10 min at room temperature. We discarded the supernatant, then washed the pellet twice with 500 uL 70% ethanol. Each wash included a 5 min incubation and a 30 s centrifugation at 13,000×g. After letting the residual ethanol evaporate for approximately 5 min, we added 25 uL low TE buffer and incubated the samples overnight to allow thorough resuspension of the pellet.
Mitochondrial DNA sequencing
We sequenced the mtDNA ND2 locus to allow for a direct comparison between our results and those of the Vredenburg et al. (2007) study. We followed the PCR protocol in Vredenburg et al. (2007) and used the conventional Sanger sequencing platform at the UC Berkeley DNA Sequencing Facility. Using the sequence analysis software Geneious, we quality trimmed the raw read data and aligned the sequences with the MUSCLE algorithm (Edgar 2004; Kearse et al. 2012). After removing low quality sequences, we analyzed this 941 bp marker in 106 individuals spread across the 23 sampling sites. After removing duplicate haplotypes present within sampling sites, we inferred the phylogeny of 41 haplotypes using a maximum likelihood approach in the program RAXML (Stamatakis 2006). We used the “GTRCAT” model of sequence evolution, identified the optimal tree from 20 separate ML searches, and conducted 500 bootstrapping replicates to evaluate node support (parameters: ‘−f d −b 500’). We used three reference sequences from Genbank for the outgroup (AF314027, AF314029, AF314030), which includes one frog from Sixty Lakes Basin in Kings Canyon National Park (R. muscosa) and two frogs from southern California Transverse Range localities (R. muscosa).
Nuclear genetic marker development
We designed primers for 50 nuclear markers from a de novo transcriptome assembly of Rana muscosa/sierrae. The markers primarily included 3′ terminal exons and flanking 3′ untranslated regions (UTR). We targeted UTR regions because they tend to be relatively variable and long in length. We began by generating de novo contigs (putative transcripts) using the mira assembler (Chevreux et al. 2004) from RNAseq 454 next-generation sequencing reads previously generated by Rosenblum et al. (2012). To identify 3′ UTR sequences in R. sierrae contigs, we followed a previously developed approach where exon alignments are generated from reference genomes (Zieliński et al. 2014). We identified exon–intron boundaries by aligning R. sierrae contigs to two reference transcriptomes: Xenopus tropicalis (Ensembl assembly v. 4.2) and Anolis carolensis (Ensembl assembly v. 2.0). We downloaded reference transcripts for X. tropicalis and A. carolensis from Biomart using the “cDNA” setting with 500 bp of 3′ downstream flanking sequence (Smedley et al. 2009). We used the blastclust program to generate alignments using the following parameters: 70% identity threshold, 50% overlap length, require coverage on both neighbors = FALSE. The resulting sequence alignment clusters were parsed in R and prepared for importation into Geneious (Kearse et al. 2012). We prioritized alignment clusters with single hits in each reference genome to limit selection of genes prone to paralogy. To prepare Xenopus and Anolis transcript annotation information for these clusters, the gene annotation files (in “gtf” format) were downloaded from Ensembl and parsed in R for importation into Geneious. Working in Geneious, we selected R. sierrae contigs with either a long exon or a long 3′ UTR (>300 bp) on a cluster-by-cluster basis. The exon–intron boundaries were mapped onto each cluster alignment using annotation information from the reference sequences. The final set of markers included 50 genomic markers (Supplementary Table 2). We then designed primers according to the manual for the target amplification platform (Fluidigm Access Array).
Nuclear DNA sequencing approach
Our sequencing approach utilized microfluidic PCR amplification of genomic markers (Fluidigm Access Array 48.48) and next-generation sequencing (Illumina Miseq). The main advantages of the Access Array platform are the high throughput amplification of markers in 48 samples (in isolated microfluidic chambers) and the production of a sequencer-ready DNA library during the marker amplification, which includes simultaneous sequencing adapter incorporation and barcode tagging of each amplicon. Access Array amplification and sequencing were performed at the University of Idaho IBEST Genomic Resources Core. To enrich the targeted regions prior to microfluidic PCR in the Access Array, we performed preamplification reactions for each sample according to manufacturer’s protocol with slight modification. Briefly, three modifications included using untagged primers (lacking “common sequence” tags used for incorporating adapter), maintaining the annealing temperature at 60 °C for all cycles, and adding one cycle to the PCR preamplification thermocycling protocol. Preamplification products were then cleaned with Exosap-it and diluted 1:5 in accordance with the manufacturer’s protocol. We then used the “preamplified” samples for Access Array amplification and sequencing. Given that the Access Array platform carries out PCR amplifications in a microfluidic grid of 48 samples x 48 primer pair pools, our primer pooling scheme included 2 2-plex primer pools and 46 1-plex primer pools. For the Illumina Miseq sequencing run, we used 2 × 300 bp paired-end reads from ¼ of the sequencing plate (~4.5 million reads), which was sufficient to generate ~470X coverage for each unique amplicon (includes every combination of sample and marker). Although this sequencing coverage might be excessive in many applications, given the variable quality of our samples we expected that the high number of reads used would minimize per-sample missing data.
Raw sequence processing and variant calling
The bioinformatics pipeline starting from raw sequencing reads included adapter and primer trimming, variant calling, and variant filtering and phasing. We used the dbcAmplicons software (https://github.com/msettles/dbcAmplicons) for trimming adapter and primer sequences from the raw data. Downstream bioinformatics steps through variant calling were performed for each sample. The paired-end reads were merged to yield extended reads that spanned the length of the marker using the flash2 software (Magoc and Salzberg 2011). We used bwa software (“mem” mode) to align the reads to the reference targeted regions (Li and Durbin 2009). We then followed the GATK ver. 3.4 best practices pipeline for calling variants (SNPs and INDELs) (Van der Auwera et al. 2013). The HaplotypeCaller tool in GATK calls SNPs and INDELs using local re-assembly of haplotypes for each sample. We then merged variant calls across all samples with the GenotypeGVCFs tool in GATK, and performed several levels of filtering to the raw variant calls. Our downstream filtering and analyses included the SNP calls only. We filtered SNP sites using standard quality control parameters (BaseQRanksSum ≤ 5, MQRankSum ≤ 3, ReadPosRankSum ≤ 4, AlleleNumber < 200). Next we removed six markers that contained two types of evidence for paralogy: (1) an excess in heterozygous calls across the majority samples and (2) an excess of mapped reads across the majority of samples. Additionally one marker was removed due to low PCR success rate (>10X coverage in only 46% of samples). The remaining 43 markers used in downstream analyses are specified in Supplementary Table 2. To determine phase of the variants, we used the ReadBackedPhasing tool in GATK for each individual. We removed samples from downstream analyses that contained a high proportion of missing data (>50%), which left 164 samples in the dataset for downstream analyses. We removed singleton alleles and used the phased haplotypes encoded as alleles in downstream analyses.
Genetic clustering
We first investigated genetic variation and clustering among samples using principal components analysis (PCA) and the Bayesian clustering method STRUCTURE. We used the adegenet R package to compute principal component (PC) scores (Jombart 2008) and then plotted the first two PCs with sampling locations used as labels. Missing genotypes were changed to the mean genotype per locus. Next we conducted an analysis using STRUCTURE (ver. 2.3.4), a multi-locus clustering program, to infer the presence of distinct populations in the dataset (Pritchard et al. 2000). This program uses a model to cluster individuals into populations that are in Hardy–Weinberg equilibrium and allows for admixture within individuals. We used Evanno’s delta K method to select the best value of number of populations (K) with the program StructureHarvester (Evanno et al. 2005; Earl and vonHoldt 2012). We ran the program under the admixture model ten times for each value of K = {2, 3, 4, 5, 6}. Each run included 20,000 MCMC steps with 10,000 steps as burn-in. Using the best value of K, we ran five long runs to assess population assignments and admixture estimates with 100,000 MCMC steps with 10,000 steps as burn-in. The results were summarized and plotted using the Clumpak program (Kopelman et al. 2015).
Spatial analysis of genetic variation
We used spatial PCA (sPCA) to analyze between-individual genetic variability in a spatially-explicit context (Jombart et al. 2008). We utilized the sPCA pipeline in the adegenet R package in order to identify spatial autocorrelation in allele frequencies in a georeferenced dataset (Jombart 2008). We used the Euclidean distance method to create a network map used to define distances between sampling locations. Following Jombart et al. (2008), we first tested for spatial autocorrelation in conventional PCA scores using Moran’s I test. Next we used the sPCA framework to conduct global (e.g. clines or patches) and local (e.g. differentiation between neighbors) tests and examined significant results by mapping spatial eigenvectors on to the geographic map. We then analyzed the hierarchical partitioning of genetic variation using spatial AMOVA, as implemented for multi-locus datasets in the spads program (Dellicour and Mardulyn 2013). We defined hierarchical levels by sampling sites and the clusters inferred from STRUCTURE using K = 3.
We performed a landscape genetics analysis at the individual level using the PopGenReport R package (Gruber and Adamack 2015). We used a raster image of elevation of the study area as a friction map to build least-cost path network between sample collection sites, which represented ecological-based distances. Grid cells of the friction map contained average elevation values (in meters) at 30 m resolution. We tested different values of the “number of neighbors” parameter in the least cost path calculation (nn = 4, 8, 16) and selected to use nn = 8 in the final analysis based on visual inspection of the path network. In the statistical analysis, we compared the predictive ability of the elevation-based cost distances and the Euclidean distances (straight-line) in explaining genetic distances among individuals using a Partial Mantel test. We opted not to test for the effects of other fine-scale ecological predictors, such as land cover, given that the genetic clustering analyses showed immediate levels of clustering by sampling locations, where samples tended to group with other samples in nearby locations, but not tight clustering by location, which would be prerequisite to fine-scale analyses.
Isolation by distance test
We tested for Isolation by distance (IBD) in the dataset, which is a positive relationship between geographic distance and genetic differentiation for pairwise comparisons of populations. We used linearized Fst (i.e. 1/(1-Fst)) as the genetic differentiation measure for sampling location. Fst was estimated using the multilocus weighted Fst method implemented in the adegenet R package (Jombart 2008). We used a Mantel test to assess the relationship between genetic and geographic pairwise distance matrices, using 10,000 permutations to estimate the significance value. Furthermore using a Partial Mantel test, we tested for the effect of a barrier between the two drainages in Yosemite: Tuolumne River drainage and Merced River drainage. The potential barrier is formed largely by the Cathedral Range ridgeline. We also used a Partial Mantel test to separate the effect of Euclidean distance from the population assignments, based on STRUCTURE results. In addition, we used multiple matrix regression (MMR) with permutation tests as another method to assess the effects of geography, drainage, and population assignment on genetic distance (Wang 2013). We repeated the regression analysis with within-drainage and between-drainage population comparisons in separate models in order to test for differences in IBD trends that may reflect the presence of a barrier between drainages. Partial Mantel and MMR computations were performed using functions in the PopGenReport R package (Adamack and Gruber 2014). To alleviate problematic statistical properties of the Partial Mantel test (Legendre and Fortin 2010), we applied causal modeling diagnostic tests (Cushman et al. 2013) implemented in the PopGenReport R package. Finally, we constructed a neighbor-joining tree based on pairwise Fst to graphically illustrate genetic differentiation patterns among populations with the phangorn package in R (Schliep 2011). Node support values were generated from 1000 bootstrap replicates.
Results
Mitochondrial DNA analysis
Our mitochondrial analyses demonstrated that samples from Yosemite were monophyletic with respect to the outgroup samples (Fig. 2, 100% bootstrap support). We found high support (98%) for monophyly of samples from the Merced River drainage with some geographic substructure but low support (<50%) for monophyly of the Tuolumne River drainage. Although the general pattern in the maximum likelihood tree was two drainage-specific haplotype groups, two sites violated this pattern. Sampled individuals from Site 11 (Unicorn Lake) carried Merced-specific haplotypes despite being situated in the Tuolumne drainage. Our sampling included haplotypes from 6 individuals with 1 haplotype represented in the tree (due to identical haplotypes among individuals). The other exception involved Site 15 (near Gallison Lake) where the reverse discordant pattern occurred: some individuals located in the Merced drainage carried Tuolumne-specific haplotypes. In this case, there was a mixture of haplotypes, in which 15 out of 17 individuals carried Tuolumne haplotypes. We note that this set of samples combined two surveys: in 2006, four out of six samples carried Tuolumne haplotypes; in 2014, 11 out of 11 carried Tuolumne haplotypes. Considering these two exceptions suggests that between-drainage migration occurs (or occurred) at least occasionally near the drainage boundary. These observations provided further motivation for assessing genetic structure with multi-locus nuclear markers. In our sampling scheme for the nuclear genetic sequencing, we included higher sample sizes when possible for sites near the watershed boundary to increase the chance of detecting between drainage connectivity.

Consensus phylogenetic tree inferred from mtDNA locus ND2. Duplicate haplotypes within populations were excluded from the analysis. Shaded bars indicate drainage location of individuals: light grey Tuolumne, dark grey Merced. Terminal nodes show respective site label of individuals. Asterisks indicate drainage-discordant haplotypes. Bootstrap support values above 50% are shown
Nuclear genetic clustering
The nuclear SNP dataset included 1251 SNPs across 43 markers after variant filtering with an average of 29 ± 16 (mean ± sd) SNPs per contig; and after removing singletons 20 ± 10 SNPs per contig. The marker lengths spanned 287–465 bp with an average of 375 ± 49 bp (mean ± sd). The results of the PCA and STRUCTURE analyses show similar patterns (Figs. 3, 4). Primarily, both analyses suggest that three clusters occur in the nuclear genetic dataset. The PCA plot shows a general pattern of clustering: sampling sites 1–7 (Tuolumne), 8–16 (Central), 17–23 (Merced) (Fig. 3). In addition, the most notable pattern in the PCA plot is that the two-dimensional summary of the genetic dataset generally mirrors the geography of the sampling locations, which is a pattern observed in other genetic studies (Novembre et al. 2008). In Fig. 3, we plotted PC1 on the y-axis and PC2 on the x-axis to facilitate visual comparison to the sample map in Fig. 1. We also note that PC1, which contains the highest proportion of variance in the dataset (by definition), generally separates individuals by drainage (Tuolumne: PC1 > -0.5; Merced: PC1 < -0.5) and strongly aligns with latitude. Given the orthogonal nature of PC axes, PC2 therefore aligns with longitude. While PC scores can reflect demographic histories (e.g. less migration along PC1), we note that previous studies have shown that PCA may artificially separate subpopulations when sampling is not spatially uniform (Novembre and Stephens 2008). This phenomenon is especially relevant for interpreting the PCA results of the Central cluster. Although PC1 shows separation of sampling sites by drainage within this cluster, we refrain from inferring demographic processes from the results (e.g. reduced migration due to a barrier) given the discontinuous nature of the habitat and therefore sampling scheme.

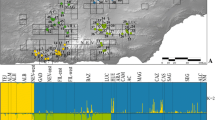
Principal components analysis (PCA) for multi-locus nucDNA, showing that individual frogs tend to cluster together according to sampling location (number inside point). Note that the plot was rotated for easier comparison to the study area: PC1 is on the y-axis and PC2 is on the x-axis. Colors correspond to proportion of ancestry for the 3 clusters from the STRUCTURE results for K = 3

STRUCTURE cluster assignment results based on multi-locus nucDNA for two, three, and four clusters (K). Each bar represents an individual frog, and colors represent inferred population ancestry. K = 3 was chosen as the best value of K by Evanno’s method
The results of the STRUCTURE analysis also indicate that three clusters occur in the genetic dataset. Using Evanno’s delta K method, the value of K = 3 was best supported by the data (supplementary Fig. 1). We show the results for different values of K={2, 3, 4}, but describe the biogeographic patterns for K = 3 (Fig. 4). Individuals from sites 1–7 (in numerical order) clustered together under the model used by STRUCTURE. These sites cluster geographically in the northwestern and northern regions of Yosemite in different tributaries of the Tuolumne drainage. Most samples in sites 8–16 clustered together and are located in the northeastern and eastern regions of Yosemite. Also situated in the eastern region, sites 17–19 share ancestry with sites 8–16, but also contain ancestry from the southern cluster: sites 18–23. We note that sites 1–14 are located in the Tuolumne drainage and sites 15–23 are located in the Merced drainage. Pairwise Fst for the three clusters were: sites 1–7 (Tuolumne) vs. sites 18–23 (Merced): Fst = 0.248; sites 1–7 (Tuolumne) vs. sites 8–16 (Central): Fst = 0.0901; sites 8–16 (Central) vs sites 18–23 (Merced): Fst = 0.0986.
Although STRUCTURE indicates that a model with three genetic clusters provides the best fit to the data, we note that we cautiously interpret the results of this analysis pending our subsequent analyses described below. Previous research suggests that genetic patterns generated by isolation by distance processes can lead to binning individuals into clusters when the actual population history is the result of more continuous processes (e.g. stepping stone model) (Guillot et al. 2009). Indeed, the K = 2 plot is suggestive of a gradient in ancestry proportions through the sampling sites. Our downstream analyses evaluate if these partitions in genetic variation are in fact artifacts (and ultimately corroborate that genetic variation is partitioned beyond a simple isolation by distance pattern).
Spatial analysis of genetic variation
The results of the spatial PCA (sPCA) indicated significant “global” spatial structure in dataset. Using the Moran’s I method, the conventional PCA score identified significant spatial autocorrelations on PC1 and PC2 (p < 0.001; p = 0.007, respectively). Given this, we retained the first two positive eigenvectors from sPCA output. The global test of the sPCA results revealed a significant positive spatial autocorrelation (p < 0.001), which is suggestive of clines or patches in the dataset. Figure 5a shows an interpolated map of sPC1 scores where the first eigenvector separates the Tuolumne and Merced sites, supporting the conventional PCA above. Likewise the sPC2 scores separate the Central cluster of sites from the Southern and Northern clusters (Fig. 5b), also similar to the conventional PCA. These results suggest that the genetic dataset is spatially partitioned along two axes, as identified in the conventional PCA, with stronger differentiation in allele frequencies between the northern Tuolumne and southern Merced sampling locations than along the border of the two drainages. Notably, the sPCA results did not identify a significant pattern of strong differentiation between neighboring sites, which is termed “local” structure in this framework (p = 0.3). Barriers would tend to create local structure due to stronger than expected differentiation between neighboring sites bisected by the barrier. As such, our a priori hypothesis regarding the Cathedral Range as a strong local barrier to migration is not supported by these results. Although sPC1 roughly divides the sampling locations into two drainages, the location of the y-axis intercept could be the combined result of the discontinuous sampling scheme and a general isolation by distance pattern. In addition, we repeated the analysis with only the Central cluster of sites. We did this to test if the spatial partitioning occurs at a finer scale and remove the strong signal in the dataset from the differentiation of individuals at the extremes of the sampling scheme (northern and southern sampling locations). Moran’s I test indicates significant positive spatial autocorrelation (p = 0.005), where sPC1 scores contain some but not complete partitioning by drainage (Fig. 5c). The global and local tests for spatial autocorrelation yielded non-significant results. While this analysis does not provide support for the strong barrier hypothesis, it remains a possibility that there are undetected, location-specific barrier(s) in this region.

Interpolated maps of spatial PCA scores based on multi-locus nucDNA for the a first and b second eigenvalues projected on the study area. Sampling locations are shown with open circles and numbers indicate locations of sample groupings. c Interpolated map of sPC1 for the subset of sites in Central region (7–19)
The results of the spatial AMOVA test largely agreed with the STRUCTURE cluster assignments when K is set at 3, where the AMOVA cluster assignments were: cluster 1 contains sites 1–7; cluster 2 contains sites 8–19; and cluster 3 contains 20–23. The highest proportion of variance occurred among populations in different clusters (Φst = 0.41). The other partitions also contain substantial amount of the variance: among populations within the same cluster: Φsc = 0.20; and among clusters: Φct = 0.26. This suggests that a substantial amount of genetic variation occurs within clusters as well as among clusters.
The landscape genetics analysis suggested that elevation-based cost distances did not better explain population structure based on our markers than the Euclidian (straight-line) distances. The partial Mantel test between genetic distance and Euclidean distance was significant when controlled for elevation-based distance (r = 0.098, p = 0.046), whereas the correlation between genetic distance and elevation-based distance was not significant when controlled for Euclidean distance (r = −0.022, p = 0.667).
Isolation by distance
We found evidence for isolation by distance not only in the spatial PCA (described above), but also from population level analyses. Fst, which is a measure of population differentiation, was significantly associated with Euclidean geographic distance (Mantel test: r = 0.632; p < 0.001; Fig. 6a). When geographic distance was controlled for using a Partial Mantel test, the three cluster assignment was a significant predictor variable of Fst (r = 0.23, p = 0.0062), which supports the three-cluster scenario predicted by STRUCTURE. Drainage (Tuolumne vs. Merced) was also a statistically significant predictor of Fst when controlling for Euclidean distance (r = 0.417, p < 0.001). In addition, the regression of Fst on distance was stronger for between-drainage pairs than for within-drainage pairs (Fig. 6b). These measures suggest that the divide between the Tuolumne and Merced drainages, including the Cathedral Range, has reduced gene flow between drainages, although the patterns do not appear indicative of a strong impermeable barrier. We also constructed a neighbor-joining tree with the pairwise Fst values to assess the branching patterns of population units at the level of sampling sites (Fig. 7). The dominant pattern in the unrooted NJ tree by visual inspection was akin to a stepping stone model of branching, but with some degree of partitioning.

a Regression of multi-locus nucDNA linearized Fst vs geographic distance for all pairwise population comparisons, showing a pattern of isolation by distance. b Regression of linearized Fst vs. geographic distance split by drainage showing a stronger IBD relationship for among-drainage pairs of populations. Among drainage population comparisons are shown in grey and within drainage comparisons are shown in black

Neighbor-joining tree based on multi-locus nucDNA pairwise Fst (linearized) of sample groupings showing some clustering of populations within drainages and some evidence for a general stepping stone pattern of divergence. Shaded boxes indicate drainage location of population: grey Tuolumne; white Merced. Bootstrap support percentages above 50% are shown
Discussion
Our analysis of genetic variation from minimally-invasive swab samples refines our understanding of population structure of the endangered R. sierrae in Yosemite National Park. Our mitochondrial dataset corroborates previous single-marker inference with samples clustering largely into two major drainages (Tuolumne and Merced clusters). Our nuclear dataset provides a more nuanced perspective suggesting that three groupings better describe population structure of this species in Yosemite National Park (Tuolumne, Central, and Merced clusters). The nuclear dataset also recovers a strong isolation by distance pattern, but the genetic variation partitions into clusters even after controlling for isolation by distance. We discuss these findings and their management implications in more detail below.
Implications of genetic variation in mtDNA
Our mtDNA results bring a clearer picture of haplotype variation among R. sierrae populations in Yosemite. First we confirmed that two major groups of haplotypes occur among the populations in Yosemite as was suggested by Vredenburg et al. (2007) (Fig. 2). The two groups are generally divided into two drainages (Tuolumne and Merced), but with two exceptions for populations located near the boundary between the two drainages. These results suggest that migration of at least female individuals occurs (or occurred) in this region near the drainage boundary. Prior to population declines over the last century, connectivity between drainages may have been more plausible than today, given the large R. sierrae population sizes in Yosemite (Grinnell and Storer 1924).
Implications of genetic variation in nuclear DNA
Our multi-locus nuclear DNA analyses provide a more refined and complex picture than the mtDNA dataset, and we highlight three key findings. The first key finding is that genetic variation among sampling locations strongly reflects geography (Figs. 3, 5). Both types of PCA (conventional and spatially-explicit) show a pattern that resemble the actual geographic locations of the samples. The PCA plots suggest that there is a semi-continuous pattern of genetic differentiation among sampling locations, rather than the strong discontinuous pattern between the two drainages as seen in the mtDNA dataset. The primary axis of genetic differentiation is nearly aligned with the North–South axis (Fig. 5a). The patterns we observe in Yosemite align with the general pattern throughout the range of R. sierrae and R. muscosa, where clade splitting occurs along the axis of the Sierra Nevada mountain range and south into the Transverse Range of southern California (Vredenburg et al. 2007). We note that this north–south axis of differentiation is correlated with the between-drainage differentiation, which is discussed in more detail below. The secondary axis, with an east–west orientation, also shows a semi-continuous pattern of differentiation among sampling locations. The patterns revealed in the PCA analyses are concordant with two additional population level analyses. The isolation by distance analysis shows a significant relationship between genetic differentiation and geographic distance between pairs of sampling locations (Fig. 6). Likewise, the neighbor-joining tree of pairwise Fst values shows the sampling locations in an approximate stepping-stone pattern (Fig. 7) and therefore supports the semi-continuous nature of differentiation (Kalinowski 2009). The signature of isolation by distance was also detected in the North American congener montane species Rana cascadae (Monsen and Blouin 2004) and among some populations of Rana pretiosa (Blouin et al. 2010), indicating some propensity for low levels of gene flow across larger geographic distances for related species.
The second key finding is that there are three main clusters in the nuclear dataset under the model in STRUCTURE (supplementary Fig. 1). The STRUCTURE clustering analysis (Fig. 4) showed that a model with K = 3 provided the best fit for the dataset. As expected given the PCA results, the clusters generally reflect the geographic structure of sampling locations. Interestingly, the Central Cluster included sampling locations from both drainages, which supports the hypothesis of ongoing or relatively recent connectivity among populations located along the drainage boundary, as suggested by the mtDNA results. The three-cluster scenario is supported by the spatial PCA analysis where the two significant axes of spatial autocorrelation (north–south and east–west) suggest that the Central cluster contained significant differentiation (Fig. 5). In addition, the three-cluster scenario is supported by the Partial Mantel test, which showed a significant correlation between genetic distance and cluster assignment when geographic distance was controlled for. The STRUCTURE cluster assignments also included substantial amounts of admixture at several sites (Fig. 4), suggesting that there is some genetic connectivity across sampling locations.
The third key finding relates to the potential role of the divide between the Tuolumne and the Merced drainages (including the Cathedral Range) as a barrier to migration, which could contribute to genetic separation among locations in different drainages. Our results suggest that this divide has a generally weak effect on migration. The fact that the Central cluster contains sampling locations on both the Tuolumne and Merced sides of the Cathedral Range suggests that this potential barrier is weak enough to allow connectivity (Fig. 5). However, our isolation by distance analysis indicate that the among-drainage pairs of locations tend to have higher differentiation after controlling for geographic distance (Fig. 6b). Considering these two analyses suggests that there are effective barriers to gene flow between certain population pairs but that some degree of historical or ongoing gene flow occurs in this system.
Previous work on the Yosemite toad (Bufo canorus), another anuran in Yosemite, provides additional context for our results. An analysis of population structure in the Yosemite toad found some similar patterns as in our study (Shaffer et al. 2000). There was a general pattern of genetic variation partitioned between Tuolumne and Merced River drainages with some between-drainage haplotype sharing in populations near the Cathedral range. A significant pattern of isolation by distance was also observed among the populations sampled across Yosemite. These patterns suggest that commonalities occur in biogeographic histories between B. canorus and R. sierrae. A likely driver for biogeography patterns is the Pleistocene glaciation events, which has been proposed for several other clades found in the Sierra Nevada (e.g. Rovito 2010; Schoville et al. 2012).
Reconciling mitochondrial-nuclear discordance across drainages
Both mitochondrial and nuclear datasets showed some degree of genetic partitioning between R. sierrae in the Merced and Tuolumne River drainages in Yosemite National Park. However, our analysis also revealed some mitochondrial-nuclear discordance. The mtDNA haplotype distribution suggested a two cluster structure as previously reported (Vredenburg et al. 2007) while the multi-locus nuclear DNA analysis suggested a three cluster structure and isolation by distance. Several processes could explain the discordance between nuclear and mitochondrial patterns at these populations including: human-assisted introductions; asymmetric dispersal, mating, or offspring production; adaptive introgression of mtDNA; and hybrid zone movement (reviewed in Toews and Brelsford 2012). Mitochondrial-nuclear discordance has been detected in other ranids (examples in Toews and Brelsford 2012). For example, discordance among datasets in Rana cascadae (a montane relative of R. sierrae) from the northwestern United States may be explained by hybrid zone movement and small population stochasticity (Monsen and Blouin 2003).
A similar scenario may have occurred in the population history of Yosemite R. sierrae, where mtDNA lineages may have evolved in isolation during glacial periods when populations retreated to lower elevations. The expected fourfold smaller effective population size of mtDNA (relative to nuclear DNA) would accelerate evolutionary divergence of mtDNA haplotypes between isolated populations. Re-colonization of high elevation habitat since the last glacial maximum with mixing of mtDNA haplotypes near the crest of the Cathedral Range may have then contributed to the observed pattern of population structure. In our dataset, we observed an unexpected distribution of mitochondrial haplotypes at two sites across the Cathedral Range (Fig. 2, Site 11, Unicorn Lake; and Site 15, Gallison Lake area). While human-assisted introduction is conceivable where frogs or tadpoles may have hitchhiked with trout during fish transplant efforts in small containers (e.g. coffee cans), our results do not support this scenario. In the “coffee can” scenario we would expect to find that recently introduced frogs would have nuclear DNA haplotypes closely related to a particular population on the opposite side of Cathedral Range, along with a high degree of admixture. Instead we find in our multi-locus nuclear analysis that frogs from Gallison and Unicorn group more closely with frogs collected at geographically proximate sites in their respective drainages (Fig. 3) with only a relatively low to moderate degree of admixture (Fig. 4). Thus our nuclear DNA results are more consistent with the scenario described above where migration occurred in the more distant past during the current interglacial period.
Minimally-invasive sampling approach
Minimally-invasive sampling techniques are especially critical for conservation studies involving threatened or endangered species. The present study represents an advance in the use of swabs from previously conducted epidemiological surveys (chytridiomycosis) for studying host population genetics. By using archived samples (including some from sampling locations that suffered subsequent population collapses) and developing new nuclear markers, we were able to conduct a fine-scale analysis of population structure in an endangered species. This approach will likely be useful for future studies on host genetics of other amphibian species given the vast number of swab-based chytridiomycosis surveys over the last decades. Genotyping from swabs allows researchers to study host population genetics without needing to re-survey populations, thus minimizing harm to study subjects. In addition, researchers can use previously collected swabs to study the genetics of extirpated populations that collapsed due to an epidemic. Thus swab archives like those used in this study represent an invaluable resource for host population genomics.
Implications for management
One of the primary tools currently being used to recover R. sierrae is the translocation of frogs from sites where they are persisting despite ongoing chytridiomycosis to habitats from which they have been extirpated (Knapp et al. 2011). Given the widespread distribution of chytridiomycosis across Yosemite (Knapp et al. 2011), pathogen transmission between sites is not a major consideration during translocations. In Yosemite, the goal of this effort is to expand the number of R. sierrae populations while preserving the historical landscape genetic structure to the extent possible. As such, gaining a better understanding of the spatial genetic structure is important for conservation management of the remaining populations. For example, in the case of translocating individuals, the preferred source population for a particular recipient population can be selected based on relatively high historical gene flow and/or short divergence time. This will minimize the primary genetic risks of translocations: outbreeding depression and loss of locally adapted alleles. Our first key finding—geographic structure with isolation by distance—suggests that translocations between neighboring geographic locations would be preferred over long-distance translocations. Our second key finding—the three cluster scenario—suggests that translocation within-clusters would be preferred over between-cluster translocations. For the “Central” cluster, a conservative approach would be to translocate individuals within drainage given the broad pattern of mtDNA partitioning by drainage and evidence of weak substructure in nuclear DNA results among sampling locations. In general, the patterns of isolation by distance and substantial within-drainage admixture suggest that using frogs from several nearby, within-cluster populations in translocations could be a reasonable strategy to re-establish extirpated R. sierrae populations that retain as much of the regional genetic diversity as possible.
References
Adamack AT, Gruber B (2014) PopGenReport: simplifying basic population genetic analyses in R. Methods Ecol Evol 5:384–387. doi:10.1111/2041-210X.12158
Blouin MS, Phillipsen IC, Monsen KJ (2010) Population structure and conservation genetics of the Oregon spotted frog, Rana pretiosa. Conserv Genet (2010) 11: 2179–2194. doi:10.1007/s10592-010-0104-x
Briggs CJ, Knapp RA, Vredenburg VT (2010) Enzootic and epizootic dynamics of the chytrid fungal pathogen of amphibians. Proc Natl Acad Sci USA 107:9695–9700
Chevreux B, Pfisterer T, Drescher B et al (2004) Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 14:1147–1159
Cushman S, Wasserman T, Landguth E, Shirk A (2013) Re-evaluating causal modeling with mantel tests in landscape genetics. Diversity 5:51–72
Dellicour S, Mardulyn P (2013) spads 1.0: A toolbox to perform spatial analyses on DNA sequence data sets. Mol Ecol Resour 14:647–651
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi:10.1093/nar/gkh340
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Grinnell J, Storer T (1924) Animal life in the yosemite. University of California Press, Berkeley
Gruber B, Adamack AT (2015) Landgenreport: a new R function to simplify landscape genetic analysis using resistance surface layers. Mol. Ecol Res 15:1172–1178
Guillot G, Leblois R, Coulon A, Frantz AC (2009) Statistical methods in spatial genetics. Mol Ecol 18:4734–4756
Hyatt AD, Boyle DG, Olsen V et al (2007) Diagnostic assays and sampling protocols for the detection of Batrachochytrium dendrobatidis. Dis Aquat Organ 73:175–192
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. doi:10.1093/bioinformatics/btn129
Jombart T, Devillard S, Dufour A-B, Pontier D (2008) Revealing cryptic spatial patterns in genetic variability by a new multivariate method. Heredity 101:92–103
Kalinowski ST (2009) How well do evolutionary trees describe genetic relationships among population. Heredity 102:506–513
Kearse M, Moir R, Wilson A et al (2012) Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Knapp RA (2005) Effects of nonnative fish and habitat characteristics on lentic herpetofauna in Yosemite National Park, USA. Biol Conserv 121:265–279. doi:10.1016/j.biocon.2004.05.003
Knapp RA, Matthews KR (2000) Non-native fish introductions and the decline of the mountain yellow-legged frog from within protected areas. Conserv Biol 14:428–438
Knapp RA, Briggs CJ, Smith TC, Maurer JR (2011) Nowhere to hide: impact of a temperature-sensitive amphibian pathogen along an elevation gradient in the temperate zone. Ecosphere 2:1–26
Kopelman NM, Mayzel J, Jakobsson M, et al. (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15: 1179–1191. doi:10.1111/1755-0998.12387
Legendre P, Fortin MJ (2010) Comparison of the Mantel test and alternative approaches for detecting complex multivariate relationships in the spatial analysis of genetic data. Mol. Ecol Res 10:831–844. doi:10.1111/j.1755-0998.2010.02866.x
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi:10.1093/bioinformatics/btp324
Lind AJ, Spinks PQ, Fellers GM, Shaffer HB (2011) Rangewide phylogeography and landscape genetics of the Western US endemic frogRana boylii(Ranidae): implications for the conservation of frogs and rivers. Conserv Genet 12:269–284
Magoc T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. doi:10.1093/bioinformatics/btr507
McCartney - Melstad E, Shaffer HB (2015) Amphibian molecular ecology and how it has informed conservation. Mol Ecol 24:5084–5109
Monsen KJ, Blouin MS (2003) Genetic structure in a montane ranid frog: restricted gene flow and nuclear-mitochondrial discordance. Mol Ecol 12:3275–3286
Monsen KJ, Blouin MS (2004) Extreme isolation by distance in a montane frog Rana cascadae. Conserv Genet 5:827–835. doi:10.1007/s10592-004-1981-z
Novembre J, Johnson T, Bryc K, et al. (2008) Genes mirror geography within Europe. Nature 456:98–101
Novembre J, Stephens M (2008) Interpreting principal component analyses of spatial population genetic variation. Nature Genet 40:646–649
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rosenblum EB, Poorten TJ, Settles M, Murdoch GK (2012) Only skin deep: shared genetic response to the deadly chytrid fungus in susceptible frog species. Mol Ecol 21:3110–3120. doi:10.1111/j.1365-294X.2012.05481.x
Rovito SM (2010) Lineage divergence and speciation in the Web-toed Salamanders (Plethodontidae: Hydromantes) of the Sierra Nevada, California. Mol Ecol 19:4554–4571. doi:10.1111/j.1365-294X.2010.04825.x
Schliep KP (2011) Phangorn: phylogenetic analysis in R. Bioinformatics 27:592–593. doi:10.1093/bioinformatics/btq706
Schoville SD, Roderick GK, Kavanaugh DH (2012) Testing the “Pleistocene species pump”in alpine habitats: lineage diversification of flightless ground beetles (Coleoptera: Carabidae: Nebria) in relation to altitudinal zonation. Biol J Linn Soc 107:95–111.
Semlitsch RD (2002) Critical Elements for Biologically Based Recovery Plans of Aquatic-Breeding Amphibians. Conserv Biol 16:619–629. doi:10.1046/j.1523-1739.2002.00512.x
Shaffer HB, Fellers GM, Magee A, Voss SR (2000) The genetics of amphibian declines: population substructure and molecular differentiation in the Yosemite Toad, Bufo canorus (Anura, Bufonidae) based on single-strand conformation polymorphism analysis (SSCP) and mitochondrial DNA sequence data. Mol Ecol 9:245–257. doi:10.1046/j.1365-294x.2000.00835.x
Shaffer HB, Fellers GM, Voss SR, Oliver JC, Pauly GB (2004) Species boundaries, phylogeography and conservation genetics of the red - legged frog (Rana aurora / draytonii) complex. Mol Ecol 13:2667–2677
Smedley D, Haider S, Ballester B et al (2009) BioMart–biological queries made easy. BMC Genom 10:22. doi:10.1186/1471-2164-10-22
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi:10.1093/bioinformatics/btl446
Stuart SN, Chanson JS, Cox NA, Young BE, Rodrigues AS, Fischman DL, Waller RW (2004) Status and trends of amphibian declines and extinctions worldwide. Science 306:1783–1786
Toews DPL, Brelsford A (2012) The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol 21:3907–3930. doi:10.1111/j.1365-294X.2012.05664.x
Van der Auwera GA, Carneiro MO, Hartl C, et al. (2013) From fastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinforma doi:10.1002/0471250953.bi1110s43
Vredenburg VT, Bingham R, Knapp R et al (2007) Concordant molecular and phenotypic data delineate new taxonomy and conservation priorities for the endangered mountain yellow-legged frog. J Zool 271:361–374. doi:10.1111/j.1469-7998.2006.00258.x
Vredenburg VT, Knapp RA, Tunstall TS, Briggs CJ (2010) Dynamics of an emerging disease drive large-scale amphibian population extinctions. Proc Natl Acad Sci USA 107:9689–9694. doi:10.1073/pnas.0914111107
Wang IJ (2013) Examining the full effects of landscape heterogeneity on spatial genetic variation: a multiple matrix regression approach for quantifying geographic and ecological isolation. Evol Int J org Evol 67:3403–3411
Zieliński P, Stuglik MT, Dudek K, et al. (2014) Development, validation and high-throughput analysis of sequence markers in nonmodel species. Mol Ecol Resour 14:352–360. doi:10.1111/1755-0998.12171
Acknowledgements
We thank Mary Toothman for assistance in retrieving archived samples, Jeanine Refsnider for assistance with experimental design, Rob Grasso for helpful comments on the manuscript, Patrick Kleeman and Brian Halstead for assistance with sample collection from the Summit Meadow frog population, and many field assistants, especially Tom Smith, Neil Kauffman, and Maxwell Joseph, for sample collection at the remaining sites. All sample collection (skin swabs) was led by Roland Knapp and authorized by research permits provided by Yosemite National Park and the Institutional Animal Care and Use Committee at University of California, Santa Barbara (UCSB). Funding was provided by the Yosemite Conservancy and a Valentine Eastern Sierra Reserve Graduate Student Research Grant from the University of California Natural Reserve System.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Poorten, T.J., Knapp, R.A. & Rosenblum, E.B. Population genetic structure of the endangered Sierra Nevada yellow-legged frog (Rana sierrae) in Yosemite National Park based on multi-locus nuclear data from swab samples. Conserv Genet 18, 731–744 (2017). https://doi.org/10.1007/s10592-016-0923-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-016-0923-5