
Abstract
Insights from conservation genomics have dramatically improved recovery plans for numerous endangered species. However, most taxa have yet to benefit from the full application of genomic technologies. The mountain yellow-legged frog species complex, Rana muscosa and Rana sierrae, inhabits the Sierra Nevada mountains and Transverse/Peninsular Ranges of California and Nevada. Both species have declined precipitously throughout their historical distributions. Conservation management plans outline extensive ongoing recovery efforts but are still based on the genetic structure determined primarily using a single mitochondrial sequence. Our study used two different sequencing strategies – amplicon sequencing and exome capture – to refine our understanding of the population genetics of these imperiled amphibians. We used buccal swabs, museum tissue samples, and archived skin swabs to genotype frog populations across their range. Using the amplicon sequencing and exome capture datasets separately and combined, we document five major genetic clusters. Notably, we found evidence supporting previous species boundaries within Kings Canyon National Park with some exceptions at individual sites. Though we see evidence of genetic clustering, especially in the R. muscosa clade, we also found evidence of some admixture across cluster boundaries in the R. sierrae clade, suggesting a stepping-stone model of population structure. We also find that the southern R. muscosa cluster had large runs of homozygosity and the lowest overall heterozygosity of any of the clusters, consistent with previous reports of marked declines in this area. Overall, our results clarify management unit designations across the range of an endangered species and highlight the importance of sampling the entire range of a species, even when collecting genome-scale data.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
High throughput sequencing has dramatically transformed the field of conservation genetics. However, there are still practical constraints for many taxa, such as amphibians, for which there is limited genomic sampling and which typically have large, complex genomes (McCartney-Melstad and Shaffer 2015; Shaffer et al. 2015; Weisrock et al. 2018). Additionally, financial limitations inherent to conservation-based research often necessitate tradeoffs when choosing management and research priorities (Maxwell et al. 2015). Therefore, researchers have often turned to reduced sequencing approaches that balance financial investment with the amount of data needed for the questions at hand (Allendorf et al. 2010; Supple and Shapiro 2018; Meek and Larson 2019). But how well do these reduced datasets capture the true genetic patterns across a landscape? This question remains largely untested, as studies of many species of conservation concern still rely on just a few mitochondrial gene sequences to inform management.
Delineating management units for a species of conservation concern is a critical first step when deciding which populations to prioritize and how and where animals could be moved on a landscape to repopulate or supplement existing populations (Moritz 1994). Moving animals across divergent genetic boundaries runs the risk of outbreeding depression, or reduced fitness caused by genetic incompatibilities and/or disruption of local adaptation (Lynch 1991; Frankham et al. 2011). However, human-assisted gene flow may be a useful strategy to quickly introduce genetic variation into a population to augment individual fitness – a process called genetic rescue (Ingvarsson 2001; Whiteley et al. 2015). Therefore, management actions relying on a foundational understanding of genetic groupings and investment in the genomics method that provides sufficient data is vital when identifying or updating management units. This is especially true for protected species for which conservation units often become codified in management plans.
The mountain yellow-legged frog species complex (Rana muscosa, Rana sierrae) provides a prime example of an endangered amphibian with ongoing recovery efforts that would benefit from increased genomic resolution. R. muscosa/sierrae were once abundant in montane aquatic communities of California and adjacent Nevada (Grinnell and Storer 1924; Stebbins 1985) but since the mid-twentieth century, have precipitously declined due to invasive fish (Bradford et al. 1993; Knapp and Matthews 2000; Vredenburg 2004; Knapp 2005; Knapp et al. 2007), the recently-emerged fungal pathogen Batrachochytrium dendrobatidis (Bd) (Rachowicz et al. 2006; Vredenburg et al. 2010), and wildfire associated flooding and debris flows (Backlin et al. 2013; Chambert et al. 2022). Given the loss of these species from > 90% of their historical range generally and over 98% in southern California specifically, there is an intensive focus on recovering frog populations using reintroductions (Briggs et al. 2005; Knapp et al. 2011; Backlin et al. 2013; Joseph and Knapp 2018; Rothstein et al. 2020; Hammond et al. 2021). Modelling indicates a need to greatly increase reintroduction experiments to stave off potential extirpation within southern California (Chambert et al. 2022). Many of these conservation actions have used genetics to decide which donor populations to use in recovery actions (e.g., Schoville et al. 2011).
The existing genetic framework for R. muscosa/sierrae is based on a single mitochondrial marker that described the major genetic management units across the species complex (Vredenburg et al. 2007). Recent frog population genetic work in Yosemite National Park, Sequoia and Kings-Canyon National Parks, and in southern California have shown that—when many nuclear genetic markers are used in tandem with higher spatial resolution from sampling many populations – these species contain high levels of spatial genetic structure (Schoville et al. 2011; Poorten et al. 2017; Rothstein et al. 2020). Moreover, genetic breaks inferred with multi-locus nuclear data are not always the same as those evident in mitochondrial trees. Therefore, an updated genetic framework for this species complex is critical for managing population and species recovery across the landscape. Additionally, genome scale data could provide invaluable insights into the levels of genetic diversity and inbreeding in each population and further inform conservation actions such as translocations and captive breeding efforts.
For protected amphibian species, like R. muscosa/sierrae, there are some challenges to obtaining genome-wide data. The protected status of these species’ limits collecting high-quality DNA sources (e.g. tissue samples). To address these limitations, our study used two approaches to collect genomic data: amplicon sequencing and exome capture sequencing. First, we used a microfluidic amplicon sequencing approach that was developed to successfully genotype DNA of low quality and quantity from skin swab samples (Poorten et al. 2017). Next, we sequenced a smaller set of existing tissue and buccal swab samples from across the range of this species complex using an exome capture approach. Exome capture sequencing allowed us to compare tens of thousands of genetic variants distributed across the coding regions of the genome, adding greater genomic resolution to our analyses. We assessed patterns of genetic structure and admixture among frog populations and explored patterns of genetic diversity among major conservation units. Our goal was to provide an extensive snapshot of genetic variation for the R. muscosa/sierrae species complex while comparing the utility of amplicon and exome capture sequencing methodologies to create a framework to inform conservation management decisions.
Materials and methods
Sampling and DNA extraction
For the exome capture assay, we compiled 96 samples, including 36 Rana muscosa, 58 Rana sierrae, and two Rana aurora samples used as an outgroup for downstream analyses: 54 were buccal swabs, and 42 were tissues. The Rana sierrae/muscosa samples represent 31 separate populations. Of the 42 tissue samples, 24 were sourced from UC Berkeley Museum of Vertebrate Zoology and California Academy of Natural Sciences archived frozen tissue collections, some representing extirpated populations. Buccal swab sample collection was authorized by research permits provided by NPS, USFWS, CDFW. To extract DNA from these samples we used Qiagen DNeasy Blood & Tissue kits following the manufacturer’s protocol.
For the amplicon sequencing assay, we used a readily available and minimally invasive source of DNA—archived skin swabs previously collected for Bd surveillance, which provided wide geographic sampling coverage. Unfortunately, skin swab extractions typically yield very little DNA, therefore they cannot be used with the exome capture approach which requires higher quality DNA samples. Samples were originally collected with a standardized approach, in which each individual frog was swabbed 30 times on the ventral skin surface. We compiled an initial set of 373 archived skin swab samples from 276 lake basins across the range of R. muscosa/sierrae. Lake basins, which represent frog “populations” in this system, are typically comprised of a series of interconnected lakes and streams. We sampled both named species Rana muscosa (n = 46) and Rana sierrae (n = 327). Additionally, we incorporated a subset of skin swab samples from previously published studies from Yosemite National Park (n = 21) (Poorten et al. 2017) and Sequoia and Kings-Canyon National Parks (n = 32) (Rothstein et al. 2020). DNA was extracted from swab samples using PrepMan Ultra Reagent and Qiagen DNeasy kits according to manufacturer’s protocol. Due to PCR inhibitors present in skin swab extracts, we used an isopropanol precipitation to purify DNA extracts. From this purified sample we used 1 µl of DNA per extract in amplicon preparation and sequencing.
Amplicon sample preparation and sequencing
We used 50 amplicon markers (400–600 bp in length) previously developed for Rana muscosa/sierrae and implemented a microfluidic PCR approach to recover nuclear amplicons (Poorten et al. 2017). We used Fluidigm Access Array and Juno microfluidic PCR platforms because they allow high throughput amplification to produce PCR products used in library preparation and sequencing. Because skin swabs typically have low quantities of DNA, we implemented a pre-amplification step based on manufacturer’s protocols (Fluidigm, South San Francisco, CA, USA). We used forward and reverse primers without tagged barcodes in an initial PCR step which increased success for downstream amplification of target amplicons. Following initial PCR, we applied an ExoSAP-IT treatment that removed PCR inhibitors (e.g. excess primers and unincorporated nucleases) and used a 1:5 dilution in nuclease-free water. Pre-amplified products were used in Illumina library preparation to include a barcoded tag of each amplicon and each sample. Illumina libraries were run on a MiSeq with 2 × 300 bp paired-end reads at the University of Idaho IBEST Genomics Resources Core, similar to Poorten et al. (2017) and Rothstein et al. (2020).
Exome capture design and sequencing
To compare the conclusions reached using the amplicon sequencing approach (required for our swab DNA samples) to an approach with higher genomic resolution, we designed an exome capture assay for Rana muscosa/sierrae. First, we sequenced the transcriptome using ventral, dorsal, liver, and spleen tissues from one individual R. muscosa. We extracted RNA using a Qiagen RNeasy extraction kit following manufacturers recommendations. All RNA extracts were assessed for integrity using a 2100 Agilent Bioanalyzer and had RIN values > 7. RNA extracts were sent to the QB3 Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley for standard RNAseq library preparation and paired-end 2 × 100 bp sequencing on 2/3 lane of an Illumina HiSeq 4000. Raw reads were cleaned following Bi et al. (2012) and Singhal (2013) and reads were assembled using Trinity (Grabherr et al. 2011).
Following sequencing we designed a custom Nimblegen SeqCap capture probe set as follows: The longest transcript per gene was selected and annotated against three available annotated genomes from related organisms (Nanorana parkeri, Xenopus tropicalis, and Anolis carolinensis) using blastx (Altschul et al. 1997) and Exonerate (Slater and Birney 2005). The Rana muscosa genome used in downstream analyses (NCBI GenBank assembly GCA_029206835.1, Hon et al. 2020) was not yet available during the capture design phase of this project. Fragmented transcripts that matched similar reference proteins were joined by Ns according to their blast hit positions. Resulting transcripts were combined to remove redundancies via CD-HIT-EST (Li and Godzik 2006) and CAP3 (Huang and Madan 1999). We defined coding sequences (cds) of each annotated transcript using Exonerate and specified these regions in a.bed file format. Pipelines used for transcriptome data processing and annotation are available at https://github.com/CGRL-QB3-UCBerkeley/MarkerDevelopmentPopGen. Final fasta sequences and bed coordinates were used for tiling cds regions for Nimblegen SeqCap EZ Developer Library (Roche Nimblegen Inc.). Probes were allowed up to 20 matches to the combined Nanorana parkeri, Xenopus tropicalis, and Anolis carolinensis reference genomes. The resulting probe set covered 99.72% of annotated transcripts with a total target size of 31.4 Mb across 14,508 targets.
We used this custom exome capture assay to sequence 94 R. muscosa/sierrae samples collected throughout the range of the species complex, and two Rana aurora samples. Extracted genomic DNA was sonicated with the qSonica Q800R and libraries were prepared using a Kapa Hyper Prep kit (Roche) incorporating uniquely dual indexes. The libraries were split between two capture pools, one for buccal swab DNA and the other for tissue, and 50 ng of each library was added to its respective pool based on a Qubit High Sensitivity assay (Invitrogen). Due to the large genome size of these frogs (10.2 Gb), we used additional input libraries (2100 ng for the tissue pool and 2800 ng for the buccal DNA pool), additional blocking oligos for adapters (10 and 15 µL Roche Universal Blocking Oligo respectively), and additional blockers for repetitive elements (for both captures 5 µL each Mouse Cot1, Human Cot1, and Chicken Hyblock + 15 µL Roche Developer Reagent) as compared with the published Nimblegen protocol. The two pools were then hybridized with the capture probe sets for 72 h at 47 °C. After the full hybridization and bead capture process, they were amplified with 9 cycles of enrichment PCR. Both capture pools were proportionately combined and run on a NovaSeq 6000 150PE Flow Cell S1 at the Vincent J. Coates Genomics Sequencing Lab at UC Berkeley, yielding 1092 M clusters of raw data.
Variant calling and filtering for exome capture data analysis
All raw reads for exome capture samples were filtered using fastp (Chen et al. 2018) and aligned to the Rana muscosa genome (NCBI GenBank assembly GCA_029206835.1, Hon et al. 2020) with repetitive elements masked using bwa (“mem” mode) (Li 2013). Variants were called using freebayes v1.3.5 (Garrison and Marth 2012). Targets for variant calling were defined as the regions in the assembled transcriptome and minimum coverage was set to 5. We then filtered variants using vcftools and the following conditions: –remove-indels –maf 0.03 –max-missing 1.0 –minQ 30 –min-meanDP 5 –max-meanDP 200 –minDP 5 –maxDP 200. We further trimmed the SNPs for some downstream analyses using the bcftools prune function to prune out SNPs in linkage disequilibrium (LD) (r2 > 0.6 in a 10 kb window) (Danecek et al. 2021). Additionally, we excluded samples with > 20% missing data and downsampled to include a maximum of three individuals per exact sampling locality. After filtering, our final exome capture dataset included 52 individuals and 20,840 SNPs.
Variant calling and filtering for amplicon data analysis
From raw sequence reads with primer sequences removed, we used the dbcAmplicons software (https://github.com/msettles/dbcAmplicons) to trim adapters sequences. Paired-end reads were merged and extended across the length of target amplicons using flash2 (Magoč and Salzberg 2011). We de-multiplexed sequences using reduce_amplicons.R script from the dbcAmplicons repository into raw.fastq for each sample. Fastq files included all sequences for each sample and were used for alignment, variant calling, and population genetic analyses.
We used bwa (“mem” mode) to align reads to target amplicon regions and created BAM files for each individual (Li 2013). From resulting BAM files, we filtered by read depth for each amplicon by sample and required an ≥ 5 reads per amplicon to pass filtering. All reads from amplicons that passed this depth filter were subsequently included in a new.bam file for each individual. Using filtered BAM files, we applied bcftools to call and output only variant sites for our unfiltered variant call file (VCF) (Li 2011; Danecek et al. 2021). We limited calls to only those within reference sequences for all 50 amplicons. From our raw VCF, we filtered variant sites using standard filtering parameters using vcftools (removed alignment mapping quality less than 30, supported base quality less than 20, include sites with MAF ≥ 0.02, exclude sites with 55% or more missing, and removed indels). Finally we removed individual samples that had more than 5% missing data using vcftools (Danecek et al. 2011), resulting in a final set of 74 individuals (60 Rana sierrae, 14 Rana muscosa) and 212 SNPs that passed our filtering steps.
Combining exome capture and amplicon data
To create a sample set with the most comprehensive geographic coverage, we combined the data from the amplicon and exome capture samples. To do this we used blastn to locate the genomic coordinates corresponding to the location of the 50 amplicon sequences in the reference genome (Altschul et al. 1997). We then used bedtools intersect to extract the genome-aligned exome capture reads from the area where the amplicons mapped to, plus an additional 500 bp on each end (Quinlan and Hall 2010). We converted the extracted bams to fastq files using picard (v.2.9.0) SamToFastq and aligned these extracted reads to a fasta containing reference amplicon sequences using bwa (Li and Durbin 2009; Broad Institute 2019). We then jointly called genotypes using the combined set of 74 amplicon samples and 52 exome capture samples with freebayes (v1.1.0–56) (Garrison and Marth 2012). We stipulated a minimum depth of 3 and stringent quality filters (flag -0) during variant calling. We excluded individuals with more than 50% missing data across raw SNPs then further filtered the variants using the following parameters: –maf 0.01 –max-missing 0.5 –minQ 30. This combined set of variants included 172 binary SNPs across 44 amplicons and 106 individuals (81 Rana sierrae, 25 Rana muscosa).
Genetic distance and clustering
Using our filtered VCFs, we conducted each analysis on either all datasets (amplicon, exome, combined) or a subset of the three datasets, depending on our specific questions and the required genomic resolution for each test. First, we inferred population genetic structure for the amplicon (N = 74 individuals, 212 SNPs), the exome capture (N = 52 individuals, 20,840 SNPs), and the combined amplicon and exome capture data (N = 106 individuals, 172 SNPs). We used discriminant analysis of principal components (DAPC) to find de novo genetic clusters in all three datasets. DAPC was implemented in the R package adegenet (v.2.1.5) (Jombart 2008). To assess the number of groupings we used the “find.clusters” function to approximate the ideal number of clusters among our samples. Briefly, “find.clusters” uses a k-means approach to find a given number of groups and maximize the variation between groups while simultaneously transforming data to retain principal components. To identify groups, the “find.clusters” function used increasing values of k (1–10). We identified the ideal number of clusters by looking for the place on the BIC chart where a flattening of criterion scores occurred (sometimes referred to as the “elbow” of the curve) (Jombart 2008). We then ran the function “optim.a.score” to find the optimal number of principal components (PCs) to use in the DAPC to avoid overfitting the data. We used 3 PCs in the exome capture analysis, 6 PCs in the amplicon only analysis, and 7 PCs in the combined analysis. Finally, we plotted each group assignment on a PCA calculated from the genetic data using the “glPca” function in adegenet and additionally plotted these clusters on a map using the R package maps showing original sampling location (Brownrigg 2018).
We also compared the amount of genetic differentiation across space and between inferred clusters. We assessed patterns of isolation by distance by comparing genetic distance (Hamming’s distance) to geographic distance (km) for the amplicon and exome capture data and calculated pairwise Fst between clusters using the hierfstat R package (Goudet 2005). Finally, for the exome capture dataset we used an AMOVA to test for the proportion of variance explained by major clusters and lake basins for our samples using ade4 (Excoffier et al. 1992; Thioulouse et al. 2018). We tested for statistical significance using a permutation test with 1000 replicates.
To further understand genetic clustering and patterns of relatedness between individuals we created a maximum likelihood phylogeny for the exome capture data. First, we converted vcf to sequence using the custom python script vcf2phylip (https://github.com/edgardomortiz/vcf2phylip/blob/master/vcf2phylip.py) and used RAxML to build a consensus maximum likelihood tree from 100 bootstrap replicates using rapid bootstrapping and search for the best-scoring tree (Stamatakis 2014). For this analysis we included two outgroup samples from the closely related Rana aurora to root the tree.
Spatial and non-spatial genetic structure using ConStruct
Because our data had a strong signature of isolation by distance (IBD), we used the R package ConStruct (Bradburd et al. 2018) to evaluate population genetic structure and admixture in the exome and amplicon datasets. ConStruct builds a model to account for IBD-driven decay in relatedness and only draws on spatial clustering when needed to explain membership in a group beyond IBD. We ran Construct on the 52 exome capture samples using a set of SNPs that were filtered and pruned for LD (as described above) and trimmed to have no missing data. Because of the sensitivity of ConStruct to missing data, for the amplicon dataset we filtered out individuals that had more than 5 missing SNPs (filtered dataset contained a total of 50 individuals). We ran cross-validation for ConStruct to compare across values of K and between spatial and non-spatial models. We ran the model 8 times for each number of clusters (K), from K = 1 to K = 8, with a chain length of 20,000 for each of the replicate runs. For the amplicon data we used a training proportion of 0.6 and for the exome capture data we used a training proportion of 0.9.
Runs of homozygosity (ROH) and individual heterozygosity for exome capture data
We leveraged our high density SNP data from the exome capture to quantify runs of homozygosity (ROH) in each of our identified clusters using the R package RZooRoH (Bertrand et al. 2019). This model-based method partitions the genome into ROH segments of varying age classes to provide insights into the history of inbreeding and bottlenecks in each population. Because of recombination during breeding events, the size of each ROH region is inversely related to the number of generations during which the regions can trace a common ancestor. Both inbreeding and population bottlenecks can increase the proportion of the genome classified as an ROH region (both in terms of number of individual regions and the sum of the size of all regions combined). We built a model with 10 Rk classes (2, 4, 8, 16, 32, 64, 128, 256, 512, 512) and used our SNP data that was not pruned for LD as input. The larger the Rk, the smaller the ROH region, therefore smaller Rk values are associate with larger, more recently created ROH. Since the Rk is approximately equal to two times the number of generations since the common ancestor of that class (Bertrand et al. 2019), our range of 10 classes captures ROH regions created between one and 256 generations ago. We then ran this model and evaluated the proportion of the genome in each of the classes, excluding the largest class which captured very small ROH regions that are less relevant to the recent history of the populations. We then calculated the number of ROH regions (NROH) and the sum of all ROH regions (SROH) and plotted these two values against each other. Finally, we also calculated the proportion of heterozygous SNPs for every individual using vcftools and plotted these values by genetic cluster. While all other exome capture analyses used a set of 20,840 binary SNPs that were quality filtered, had no missing data across all individuals, and were pruned for LD, for the ROH analysis we used a set of SNPs that was not pruned for LD (N = 66,367). R code for ROH analysis was modified by R. Gooley and AQB (to account for the yellow-legged frog genome size and SNP density of dataset) from code written by R. Gooley (and previously published in Coimbra et al. 2021). Resulting code can be found at: https://github.com/allie128/rana-rangewide/blob/main/RZooRoH_analysis_rana.rmd
Results
Exome capture data
Our range-wide set of exome capture samples could best be described by five major genetic clusters (Fig. 1). Geographic and genetic distances were strongly correlated for the exome capture data (Mantel r = 0.57, p < 0.0004, Fig. 2a). The Mantel correlation coefficient was positive and statistically significant for comparisons within ~ 100 km (Fig. 2b). The Bayesian Information Criterion (BIC) for successive values of K in the DAPC and the cross-validation results from the spatial ConStruct model showed minimal model improvement after K = 5 (Fig. 3c,d; Figure S1a), indicating K = 5 was the best fit for the data. The DAPC for the exome capture data used the first 3 PCs, 3 discriminant functions, and accounted for 56.5% of the variance in the data. Plotting these clusters on a PCA shows distinct groups with non-overlapping 95% confidence ellipses (Fig. 1c). As shown on the map (Fig. 1b) and in the phylogeny (Fig. 1a), there are three clusters within R. sierrae (here named “Northern R. sierrae”, “East Yosemite R. sierrae”, and “Southern R. sierrae”) and two clusters within R. muscosa (here named “Northern R. muscosa” and “Southern R. muscosa”). Fst is lowest between clusters within R. sierrae and highest between the Southern and Northern R. muscosa clusters and all other clusters (Fig. 3a). The AMOVA indicates that 47.1% of the variation in the exome capture data is explained by the K = 5 clusters (p < 0.001), 22.5% of the variation is explained at the population (= lake basin) level within clusters (p < 0.001), and 30.2% of the variation can be attributed to variation among individual samples (p < 0.001) (Figure S2).

Phylogeny and PCA biplot from exome capture data show five genetic clusters for Rana muscosa and Rana sierrae. a Phylogeny showing 52 Rana muscosa/sierrae exome capture samples, with two Rana aurora exome capture samples as outgroups. Tree calculated from 20,861 SNPs with RAxML. Node color represents bootstrap support from 100 replicates. Sample names are colored as in PCA and map. b Map of sampling locations for 52 Rana muscosa/sierrae exome capture samples. c PCA calculated from 20,840 SNPs. Colors represent groupings assigned using DAPC with K = 5. Cluster abbreviations are as follows: S RAMU Southern Rana muscosa, N RAMU Northern Rana muscosa, S RASI Southern Rana sierrae, EY RASI East Yosemite Rana sierrae, N RASI Northern Rana sierrae

Strong pattern of isolation by distance, especially within 100 km for Rana muscosa/sierrae. Plots showing pairwise genetic distance (Hamming distance as calculated using the “bitwise.dist” function in the R package poppr v.2.9.3) vs pairwise geographic distance in km for the (a) exome capture SNP dataset, and the (c) amplicon SNP dataset. Mantel correlogram for (b) exome capture samples and (d) amplicon samples. Filled in circles in the correlograms show statistically significant (p < 0.05) correlation between genetic and geographic distance at a given bin

Population differentiation and patterns of heterozygosity for the five identified Rana muscosa/sierrae clusters. a Pairwise Fst values and heatmap for each of the five clusters as identified using DAPC. Calculated from 20,840 exome capture SNPs. b Violin plot showing the proportion of heterozygous SNPs for each cluster. Statistically significant (p < 0.05) comparisons between clusters using a two-sided Wilcoxon rank sum test are shown with ***. c Barchart showing the proportion of the genome that is associated with runs of homozygosity (ROH) regions of different sizes (as indicated by colors) for each of the five major clusters for Rana muscosa/sierrae. Larger values for Rk represent smaller ROH regions while smaller values represent larger, and therefore more recently formed ROH regions. Three samples collected from the Independence population are denoted with a + . d Scatterplot showing the relationship of the number of ROH regions on the y-axis vs the sum of the size of all ROH regions on the x-axis. Symbols represent the five clusters identified using DAPC. Cluster abbreviations as in Fig. 1
The ConStruct analysis showed higher predictive accuracy for the spatial models rather than the non-spatial models for all values of K (Fig. 4c, d) as expected given the signature of isolation by distance (IBD) in the data. The spatial ConStruct model for K = 2 highlights a more dramatic shift in admixture patterns between the two species than the non-spatial model, highlighting this important genetic break (Fig. 4a, b). Both the spatial and non-spatial models at K = 5 show a pattern of gradual shifts in admixture within R. sierrae versus distinct sub-populations within R. muscosa. This pattern can also be seen in the phylogeny: the R. sierrae clade shows a pattern of stepwise branching and the R. muscosa clade shows an initial main split (Fig. 1a).

Spatial admixture models show clear species boundaries and some admixture within species. For the exome capture dataset, results from spatial (a) non-spatial (b) ConStruct models for K = 2 to K = 5. Individuals are arranged according to DAPC cluster. Vertical lines indicate where the DAPC clusters separate samples for K = 1 to K = 5, with the solid line separating the two species and the dashed lines separating clusters within species. c Cross-validation results for spatial (blue) and non-spatial (green) models for K = 1 to K = 8. d Spatial cross-validation results from (c) plotted using a more optimal y-axis range. Cluster abbreviations as in Fig. 1
Finally, to evaluate the genetic diversity of each population we quantified runs of homozygosity (ROH) and calculated the proportion of heterozygous SNPs for each individual. Here, we found an unusually high proportion of the genome classified as smaller ROH regions in the Rk class of 64–128 for all three individuals from the Independence population (Southern R. sierrae cluster; Fig. 3c). A Rk class can be thought of as a bin containing ROH regions of a certain length. We can approximate the age of the regions in generations as the Rk divided by two (Bertrand et al. 2019), or between 32 and 64 generations ago. By comparing the sum of all the ROH regions (in Mb) to the number of unique ROH regions, we see that these three individuals are outliers along both axes (Fig. 3d). Similarly, the Southern R. muscosa cluster has fewer, larger ROH regions than the rest of the clusters (Fig. 3c,d). Many of these regions fall within the Rk classes of 8 and 16, indicating these regions were created between four and eight generations ago. The Southern R. muscosa samples had the lowest proportion of heterozygous SNPs, while the East Yosemite R. sierrae cluster had the highest proportion of heterozygous SNPs. All other clusters were intermediate and not significantly different from each other (Fig. 3b).
Amplicon data
For our amplicon sequence dataset, after stringent filtering that excluded samples with more than five missing SNPs, we included a total of 74 out of the original 373 skin swab samples we attempted to sequence. While this final dataset included only 19.8% of the original samples, it still included samples from every part of the range of the species complex (Figure S3). Site level filtering yielded 212 binary SNPs across 44 nuclear amplicon markers. Generally, there was a strong pattern of IBD (Mantel r = 0.32, p < 0.0004) and the strongest correlation of genetic and geographic distance occurred within ~ 50 km (Fig. 2 c,d). Both Bayesian Information Criterion for DAPC and cross-validation results from the spatial ConStruct model showed minimal model improvement after K = 5 (Figure S1b), indicating this was the best fit for the data. PCA axes highlight a major split in the data along PC1 (28.1% of variation in the data) that split samples within Yosemite National Park. This split can be seen in the results from the DAPC at K = 2 which used the first 6 PCs, 4 discriminant functions, and conserved 63.9% of the variance in the data (Figures S3c, d). For K = 5 the DAPC from the amplicon data showed additional splits along the range. There is only one amplicon sample from the southern disjunct range of R. muscosa that passed quality filters, but notably this sample grouped with the southernmost samples of R. muscosa in the Sierra Nevada.
Amplicon and exome capture combined data
By combining the amplicon and exome capture samples, we created an intermediate dataset that had the advantage of increased sample size (N = 106) and geographic coverage. This dataset had similar genetic resolution as the amplicon-only dataset (N = 176 binary SNPs across 44 amplicons). We did not see evidence of a batch effect (i.e., samples clustering by sequencing method) in the PCA or in the DAPC clustering results (Fig. 5b). The BIC chart for the combined dataset did not show a clear inflection point across successive values of K (Figure S1c), so we investigated the relevant values of K highlighted for the separate datasets: K = 2 to explore the species boundary, and K = 5 to evaluate for clusters within species. We found that at K = 2 the combined dataset places the species boundary at the same place as the exome capture dataset, within Kings Canyon National Park (Fig. 5a). Additionally, at K = 5 the clusters identified in the combined dataset largely match the K = 5 cluster boundaries in the exome capture data. For example, the same boundaries were identified between the three R. sierrae clusters in Yosemite National Park (Fig. 6a). Pairwise Fst largely matched the results from the exome capture data, with the largest distance between the Southern R. muscosa cluster and all other populations, and smaller Fst values between populations within the R. sierrae clade.

Species boundary lies within Kings Canyon National Park. a Map of sampling locations for the combined dataset, including 55 amplicon samples (circles) and 51 exome capture samples (triangles). Colors represent clusters identified with the DAPC using K = 2 for a set of 172 binary SNPs. Insert shows a zoomed in representation of Sequoia and Kings Canyon National Parks with the South Fork of the Kings River labeled. Arrows indicate direction of flow for each river. Muro Blanco basin is highlighted with a *. b PCA with shapes and colors as in map

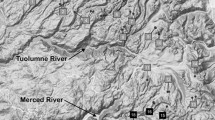
Admixture common at cluster boundaries for Rana sierrae within Yosemite National Park. a Map of sampling locations for the combined dataset, including 55 amplicon samples (circles) and 51 exome capture samples (triangles). Colors represent clusters identified with the DAPC using K = 5 for a set of 172 binary SNPs. Insert shows a zoomed in representation of Yosemite National Park with the Tuolumne River and Merced River labeled. Arrows indicate direction of flow for each river. b PCA with shapes and colors as in map. Cluster abbreviations as in Fig. 1
Discussion
In our study, we used two different sequencing and sampling strategies for the Rana muscosa/sierrae species complex and compared population genetic results using each approach. We also combined these two datasets to evaluate the influence of incomplete sampling versus limited genetic markers. For our amplicon sequencing approach, we leveraged archived skin swab samples and genotyped using a custom microfluidic PCR-based assay. We also used an exome capture sequencing approach with custom targets to genotype tissues and buccal swabs from across the species complex range, resulting in ~ 100X more high-quality genetic variants than the amplicon dataset. Each of these datasets has shortcomings: the amplicon data have very few SNPs and only include a single sample from the southern disjunct range. In contrast, the exome capture dataset has over 20,000 SNPs, but has a sampling scheme that emphasizes repeat sampling of the same populations rather than sampling all known populations. Therefore, by combining the two and calling SNPs only in the shared genomic regions present in both datasets, we can ameliorate the issue of limited sampling to see how that may have influenced the genetic clusters identified in the amplicon approach. Together, these datasets create a relatively complete genomic picture for these imperiled amphibians and allow the identification of key methodological considerations for conservation genomic studies.
Support for previous species boundaries and shifting within-species genetic groups
Previous work identified six phylogenetic groupings in R. muscosa/sierrae and named a species level split based on mitochondrial, morphometric, and acoustic data (Vredenburg et al. 2007). Our work – with vastly increased numbers of genetic markers using multiple methods – largely reflects the original boundaries of the R. muscosa/sierrae species split and suggests only minor changes to the originally identified clusters. Our amplicon data, which included more samples across many different locations, indicated that the largest genetic split occurred between samples collected from Yosemite National Park (Figure S3C). However, results from the exome capture data align more with previous studies, showing the major species split within populations in Kings Canyon (Fig. 1b). Using the combined dataset, we see genetic clusters that match those found in the exome capture analyses (Figs. 5,6), adding some additional geographic resolution to the cluster boundaries because of the increased sample size.
We confirm that the boundary between R. sierrae and R. muscosa lies between the south and middle forks of the Kings River in Kings Canyon National Park (Fig. 5). While this boundary may be similar to the location previously identified using only mitochondrial sequence data across the whole species complex (Vredenburg et al. 2007), there are differences at the local scale. For example, a sample from the Muro Blanco Basin (see * on Fig. 5a) was assigned to R. muscosa in the Vredenburg et al. (2007) study, but here is grouped with the southernmost clade of R. sierrae. Unfortunately, our sampling does not include many samples at the northernmost reach of the South Fork of the Kings River, where R. muscosa was previously documented (Vredenburg et al. 2007). Therefore, we conclude that at a gross level the major species boundary should remain unchanged, but that better sampling at the border of these two species (along the South Fork of the Kings River) may serve to further clarify this boundary. In contrast to the original study by Vredenburg et al. (2007), we found that the southern R. muscosa population is better represented by two clusters rather than three. Our data show one cluster restricted to the southern disjunct range (Transverse/Peninsular Ranges in southern California) and the other cluster extending from the southernmost populations in the Sierra Nevada north to just below the South Fork of the Kings River (Fig. 1b, Fig. 5). Fst values indicate that these clusters are strongly differentiated (Fig. 3a, S4) and differ significantly in average heterozygosity (Fig. 3b). This agrees with previous studies documenting significant genetic breaks between these two geographically distant clades (Schoville et al. 2011).
We inferred three genetic clusters within R. sierrae and found that the borders among all three can be found within Yosemite National Park. The newly identified East Yosemite clade includes samples from the headwaters of the Tuolumne and Merced rivers on the eastern side of the park (Figs. 1b, 6). The border between the Tuolumne River watershed and the Merced River watershed roughly separates the Southern and Northern R. sierrae clades, with a few exceptions (Fig. 6a). Yosemite may be the site of multiple genetic breaks because of barriers formed during Pleistocene glaciations (Swenson and Howard 2005) and subsequent post-glacial dispersal. Indeed, similar genetic patterns have been observed in the Yosemite Toad (Anaxyrus canorus), suggesting multiple Sierra Nevada amphibians species were influenced by similar forces across the landscape (Maier et al. 2019). However, admixture is likely occurring between R. sierrae clusters as there is significant geographic overlap between clusters in this area (Fig. 6a). Adding evidence in support of admixture, in R. sierrae the phylogeny shows continual branching rather than reciprocal monophyly between clades, implying a stepping-stone pattern of relatedness (Fig. 1a). Importantly, the two amplicon samples located within the borders of the East Yosemite cluster but assigned to the Norhernn R. sierrae clade have ~ 50% missing data, which could add uncertainty to their cluster assignment. Therefore, neighboring lake basins may be more closely related in this area regardless of inferred boundaries between genetic groups.
Patterns of ROH and heterozygosity reveal low genetic diversity in southern R. muscosa
Runs of homozygosity (ROH) in a genome form when an individual inherits two identical copies of a chromosomal segment from its ancestors. When closely related individuals breed, many large ROH regions form in the genome due to the combination of identical chromosome copies. Therefore, signatures of ROH, both the size and the number of ROH regions, can provide insights on possible inbreeding and/or population bottlenecks (Ceballos et al. 2018; Bertrand et al. 2019). In our ROH analysis, we first identified three outlier samples from the same population – Independence – that belong to the Northern R. sierrae clade. These three samples had an unusually high proportion of their genome classified in the Rk class of 64–128 (Fig. 3c,d), which corresponds to ROH regions created approximately 32–64 generations ago. This population is also interesting because it is the southernmost member of the Northern R. sierrae clade and extends further south and east than samples in the Southern R. sierrae clade (Fig. 5). According to the ROH results, this population may have gone through a strong bottleneck between 32 and 64 generations ago. This estimate roughly matches the timing of trout introduction (~ 150 years ago) in this area (Pister 2001), which caused a large bottleneck in the R. sierrae population (Knapp and Matthews 2000). Trout were subsequently eradicated from the site in the early 2000s as part of an effort to protect vulnerable frog populations. Our ROH analysis also found that the Southern R. muscosa clade tended to have fewer, larger ROH regions (Fig. 3c,d) dating to ~ 4–8 generations ago, perhaps coinciding with populations declines in that region (Backlin et al. 2013). Additionally, the Southern R. muscosa samples have significantly lower heterozygosity than all other clades (Fig. 3b), highlighting this clade as perhaps in need of interventions to supplement dwindling genetic diversity (Whiteley et al. 2015).
Updating management strategies to recognize new genetic boundaries and low genetic diversity
Given observed patterns of IBD, there are some clear management actions suggested from our results. Our results roughly agree with the original species boundary – with some exceptions at individual sites along the boundary line – but suggest modifications be made to genetic groups within the designated species. We observed five distinct genetic clusters with varying levels of admixture across cluster boundaries suggesting a stepping-stone model of population structure in R. sierrae and a more structured split into two clades in R. muscosa. Therefore, species should continue to be managed as separate groups and genetic clusters could be used operationally as functional conservation units. In cases of reintroductions, moving frogs within clusters may be an appropriate management strategy to preserve historical genetic structure. Such movements would also likely better maintain any locally adapted alleles. In a separate study, we found strong spatial structure of Bd in the Sierra Nevada (Rothstein et al. 2021). Therefore, restricting movement of frogs to only adjacent populations would also reduce mixing of Bd genotypes, and minimize the chances of any unforeseeable consequences.
A conservative approach to maintaining historical genetic structure may be appropriate in many cases as this maintains the historical biogeographic signal. However, in certain parts of the range, a more aggressive management strategy might be warranted, from a genetics perspective. For instance, high genetic distinctiveness and low genetic diversity in the Southern R. muscosa clade could be a warning sign of compromised genetic health of these populations (see also Peek, O’Rourke, and Miller 2021). Southern populations of R. muscosa have experienced some of the worst declines of the species complex (up to 98% of historical populations lost) and have limited options for local donors to bolster populations, which led to the development of a captive breeding program (Backlin et al. 2013). Management options for southern populations have always seemed limited because previous results suggested no historical admixture between southern frogs and the rest of the range, with three main historic sub-populations defined in this southern area. (Schoville et al. 2011).
Our study largely supports this finding; however, one of the biggest barriers to recovery of these southern frogs is the lack of suitable habitats for reintroduction experiments. Where in situ mitigation has taken place (trout removal and fish barrier installation) population recovery has been a success (see Little Rock on Fig. 3 of Chambert et al. 2022), but the recent drought has further reduced habitat suitability across all sites. So, although our data suggest that there may be an opportunity to use donor individuals from large, persistent populations in Sequoia and Kings Canyon National Parks to augment dwindling southern population genetic diversity while maintaining historical population structure, this option might be limited in the current landscape. Future investigations to assess whether translocation of frogs between these two regions is justified. Outcomes of translocation could be evaluated at the currently un-occupied site where no frogs are established at Breckinridge Mountain, which is between the northern and southern frogs. Further there is reasonable variability within the southern frogs (Fig. 1a) and interbreeding of these populations could be tried at the un-occupied Palomar Mountain at the southern edge of the range.
Comparing sequencing methods for conservation genetic projects
Collecting genome-scale data for many individuals is becoming increasingly affordable, allowing for impressive genomic and spatial resolution for conservation genetic studies. By directly comparing multi-locus (i.e., microsatellites, mtDNA), reduced representation (i.e., amplicon sequencing, RADseq), and genome-wide (i.e., exome capture, whole genome resequencing) sequencing methods, we can help integrate new sequencing data with previous studies and better contextualize the relationship between sample size, sampling design, and population genetic inferences. Here we found somewhat different genetic clusters when using amplicon-based SNP data versus exome-capture based SNP data. Perhaps most notably, the K = 2 boundary was placed in Yosemite using amplicon data but further south in Kings Canyon National Park with the exome capture data (Fig. 1, Figure S3). To investigate this difference, we combined these different data to create a dataset with similar genetic resolution as the amplicon dataset, but with more comprehensive sampling. Using the combined dataset, we found cluster boundaries matching those obtained from the exome capture dataset (Figs. 5, 6), adding confidence to our conclusions made from the exome capture data and revealing that incomplete sampling of the southern part of the range (after filtering out samples), rather than the limited number of SNPs, likely biased amplicon results.
In summary, for population genetic studies, boosting sample representation across populations may be the best strategy if scientists need to choose between increased genomic or geographic resolution. However, the opportunities for addressing previously intractable questions using genome-scale data are enormous and can satisfy needs to perform population genetic structure analyses at the same time. In this study, we used exome capture data for a focused set of research questions and such data can be applied to many more. For example, we leveraged our genome-wide SNPs and a high-quality reference genome to evaluate patterns of ROH in the genome, which would not have been possible with amplicon-based SNPs or an incomplete reference genome. Studies are underway that use these data and other whole exome sequences from these species to identify genes associated with population persistence in the face of disease for this endangered amphibian.
Conclusions
Creating a comprehensive genetic framework for conservation is crucial for declining species. Delineating historical population genetic structure and diversity, especially when current populations are vanishing, can guide and strengthen species recovery efforts. Here, we gathered a comprehensive set of samples from across the range of R. muscosa/sierrae, taking advantage of archived skin swabs, museum tissues, and buccal swabs, to investigate historical genetic population structure and diversity. We also explored the impacts that choice of sequencing technology and sampling strategy can have on population genetic inferences, finding that, when genetic markers are limited, sampling design is critical for inferring number of clusters and delimiting their boundaries. Using our robust set of ~ 20,000 exome capture SNPs we identified key genetic units across the R. muscosa/sierrae range. Our work provides a comprehensive framework to guide ongoing conservation management. We found that genetic clusters primarily exhibit a pattern of isolation by distance and that clusters are somewhat permeable to gene flow, especially for R. sierrae. Importantly, we found that some clusters (southern R. muscosa) are more genetically isolated and less genetically diverse than others, a signature that may result from a recent history of population declines. We also found evidence supporting the primary species-level split and better inform which clusters could be used as donors to support recovery efforts in neighboring clusters, which may be necessary given the evidence of inbreeding and low genetic diversity in clades such as the southern R. muscosa group. Although genetic diversity is very low in some populations, the fact that some populations persist in the face of extreme bottlenecks (see Knapp et al. 2016) is evidence that these frogs can survive, even in the absence of genetic rescue. Overall, our results create a more explicit blueprint for framing management actions for an imperiled species group and provide insights into the influence of genomic resolution and sampling design.
Data availability
All raw sequencing data are available on the NCBI SRA (PRJNA870451). All R code used for data analysis and sample metadata is available on github (https://github.com/allie128/rana-rangewide).
Change history
19 April 2024
A Correction to this paper has been published: https://doi.org/10.1007/s10592-024-01615-9
References
Allendorf FW, Hohenlohe PA, Luikart G (2010) Genomics and the future of conservation genetics. Nat Rev Genet 11:697–709. https://doi.org/10.1038/nrg2844
Altschul SF, Madden TL, Schäffer AA et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. https://doi.org/10.1093/nar/25.17.3389
Backlin AR, Hitchcock CJ, Gallegos EA et al (2013) The precarious persistence of the Endangered Sierra Madre yellow-legged frog Rana muscosa in Southern California, USA. Oryx 49:157–164. https://doi.org/10.1017/s003060531300029x
Bertrand AR, Kadri NK, Flori L et al (2019) RZooRoH: an R package to characterize individual genomic autozygosity and identify homozygous-by-descent segments. Methods Ecol Evol 10:860–866. https://doi.org/10.1111/2041-210X.13167
Bi K, Vanderpool D, Singhal S et al (2012) Transcriptome-based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales. BMC Genomics 13:403. https://doi.org/10.1186/1471-2164-13-403
Bradburd GS, Coop GM, Ralph PL (2018) Inferring continuous and discrete population genetic structure across space. Genetics. https://doi.org/10.1534/genetics.118.301333
Bradford DF, Tabatabai F, Graber DM (1993) Isolation of remaining populations of the native frog, Rana muscosa, by introduced fishes in Sequoia and Kings Canyon National Parks, California. Conserv Biol 7:882–888. https://doi.org/10.1046/j.1523-1739.1993.740882.x
Briggs CJ, Vredenburg VT, Knapp RA, Rachowicz LJ (2005) Investigating the population-level effects of chytridiomycosis: an emerging infectious disease of amphibians. Ecology 86:3149–3159. https://doi.org/10.1890/04-1428
Broad Institute (2019) Picard Toolkit. In: Github Repos. https://github.com/broadinstitute/picard.
Brownrigg R (2018) maps: Draw geographical maps. R package version 3.3. 0 (original S code by Richard A. Becker, Allan R. Wilks with enhancements by Thomas P Minka and Alex Deckmyn)
Ceballos FC, Joshi PK, Clark DW et al (2018) Runs of homozygosity: windows into population history and trait architecture. Nat Rev Genet 19:220–234. https://doi.org/10.1038/nrg.2017.109
Chambert T, Backlin AR, Gallegos E et al (2022) Defining relevant conservation targets for the endangered Southern California distinct population segment of the mountain yellow-legged frog (Rana muscosa). Conserv Sci Pract. https://doi.org/10.1111/csp2.12666
Chen S, Zhou Y, Chen Y, Gu J (2018) Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. https://doi.org/10.1093/bioinformatics/bty560
Coimbra RT, Winter S, Kumar V, Koepfli KP, Gooley RM, Dobrynin P, Fennessy J, Janke A (2021) Whole-genome analysis of giraffe supports four distinct species. Current Biology. Jul 12;31(13):2929–38. https://doi.org/10.1016/j.cub.2021.04.033
Danecek P, Auton A, Abecasis G et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Danecek P, Bonfield JK, Liddle J et al (2021) Twelve years of SAMtools and BCFtools. Gigascience. https://doi.org/10.1093/gigascience/giab008
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491. https://doi.org/10.1093/genetics/131.2.479
Frankham R, Ballou JD, Eldridge MD et al (2011) Predicting the probability of outbreeding depression. Conserv Biol 25:465–475. https://doi.org/10.1111/j.1523-1739.2011.01662.x
Garrison E, Marth G (2012) Haplotype-based variant detection from short-read sequencing. ArXiv. https://doi.org/10.48550/arXiv.1207.3907
Goudet J (2005) Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–186. https://doi.org/10.1111/j.1471-8286.2004.00828.x
Grabherr MG, Haas BJ, Yassour M et al (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652. https://doi.org/10.1038/nbt.1883
Grinnell J, Storer TI (1924) Animal life in the Yosemite: an account of the mammals, birds, reptiles, and amphibians in a cross-section of the Sierra Nevada. University of California Press
Hammond TT, Curtis MJ, Jacobs LE et al (2021) Overwinter behavior, movement, and survival in a recently reintroduced, endangered amphibian, Rana muscosa. J Nat Conserv. https://doi.org/10.1016/j.jnc.2021.126086
Hon T, Mars K, Young G et al (2020) Highly accurate long-read HiFi sequencing data for five complex genomes. Sci Data 7:1–11. https://doi.org/10.1038/s41597-020-00743-4
Huang X, Madan A (1999) CAP3: A DNA sequence assembly program. Genome Res 9:868–877. https://doi.org/10.1101/gr.9.9.868
Ingvarsson PK (2001) Restoration of genetic variation lost - The genetic rescue hypothesis. Trends Ecol Evol 16:62–63. https://doi.org/10.1016/S0169-5347(00)02065-6
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Joseph MB, Knapp RA (2018) Disease and climate effects on individuals drive post-reintroduction population dynamics of an endangered amphibian. Ecosphere. https://doi.org/10.1002/ecs2.2499
Knapp RA (2005) Effects of nonnative fish and habitat characteristics on lentic herpetofauna in Yosemite National Park, USA. Biol Conserv 121:265–279. https://doi.org/10.1016/j.biocon.2004.05.003
Knapp R, a., Boiano DM, Vredenburg VT, (2007) Removal of nonnative fish results in population expansion of a declining amphibian (mountain yellow-legged frog, Rana muscosa). Biol Conserv 135:11–20. https://doi.org/10.1016/j.biocon.2006.09.013
Knapp RA, Matthews KR (2000) Non-native fish introductions and the decline of the mountain yellow-legged frog from within protected areas. Conserv Biol 14:428–438. https://doi.org/10.1046/j.1523-1739.2000.99099.x
Knapp RA, Briggs CJ, Smith TC, Maurer JR (2011) Nowhere to hide: impact of a temperature-sensitive amphibian pathogen along an elevation gradient in the temperate zone. Ecosphere. https://doi.org/10.1890/ES11-00028.1
Knapp RA, Fellers GM, Kleeman PM et al (2016) Large-scale recovery of an endangered amphibian despite ongoing exposure to multiple stressors. Proc Natl Acad Sci 113:11889–11894. https://doi.org/10.1073/pnas.1600983113
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993. https://doi.org/10.1093/bioinformatics/btr509
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659. https://doi.org/10.1093/bioinformatics/btl158
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. https://doi.org/10.48550/arXiv.1303.3997
Lynch M (1991) The genetic interpretation of inbreeding depression and outbreeding depression. Evolution 45:622–629. https://doi.org/10.1111/j.1558-5646.1991.tb04333.x
Magoč T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. https://doi.org/10.1093/bioinformatics/btr507
Maier PA, Vandergast AG, Ostoja SM et al (2019) Pleistocene glacial cycles drove lineage diversification and fusion in the Yosemite toad (Anaxyrus canorus). Evolution (n y) 73:2476–2496. https://doi.org/10.1111/evo.13868
Maxwell SL, Rhodes JR, Runge MC et al (2015) How much is new information worth? Evaluating the financial benefit of resolving management uncertainty. J Appl Ecol 52:12–20. https://doi.org/10.1111/1365-2664.12373
McCartney-Melstad E, Shaffer HB (2015) Amphibian molecular ecology and how it has informed conservation. Mol Ecol 24:5084–5109. https://doi.org/10.1111/mec.13391
Meek MH, Larson WA (2019) The future is now: Amplicon sequencing and sequence capture usher in the conservation genomics era. Mol Ecol Resour 19:795–803. https://doi.org/10.1111/1755-0998.12998
Moritz C (1994) Defining ‘evolutionarily significant units’ for conservation. Trends Ecol Evol 9:373–375. https://doi.org/10.1016/0169-5347(94)90057-4
Peek RA, O’Rourke SM, Miller MR (2021) Flow modification associated with reduced genetic health of a river-breeding frog, Rana boylii. Ecosphere. https://doi.org/10.1002/ecs2.3496
Pister EP (2001) Wilderness fish stocking: history and perspective. Ecosystems 4:279–286. https://doi.org/10.1007/s10021-001-0010-7
Poorten TJ, Knapp RA, Rosenblum EB (2017) Population genetic structure of the endangered Sierra Nevada yellow-legged frog (Rana sierrae) in Yosemite National Park based on multi-locus nuclear data from swab samples. Conserv Genet 18:731–744. https://doi.org/10.1007/s10592-016-0923-5
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. https://doi.org/10.1093/bioinformatics/btq033
Rachowicz LJ, Knapp RA, Morgan JAT et al (2006) Emerging infectious disease as a proximate cause of amphibian mass mortality. Ecology 87:1671–1683
Rothstein AP, Knapp RA, Bradburd GS et al (2020) Stepping into the past to conserve the future: archived skin swabs from extant and extirpated populations inform genetic management of an endangered amphibian. Mol Ecol 29:2598–2611. https://doi.org/10.1111/mec.15515
Rothstein AP, Byrne AQ, Knapp RA et al (2021) Divergent regional evolutionary histories of a devastating global amphibian pathogen. Proc R Soc B. https://doi.org/10.1098/rspb.2021.0782
Schoville SD, Tustall TS, Vredenburg VT et al (2011) Conservation genetics of evolutionary lineages of the endangered mountain yellow-legged frog, Rana muscosa (Amphibia: Ranidae), in southern California. Biol Conserv 144:2031–2040. https://doi.org/10.1016/j.biocon.2011.04.025
Shaffer HB, Gidiş M, McCartney-Melstad E et al (2015) Conservation genetics and genomics of amphibians and reptiles. Annu Rev Anim Biosci 3:113–138. https://doi.org/10.1146/annurev-animal-022114-110920
Singhal S (2013) De novo transcriptomic analyses for non-model organisms: an evaluation of methods across a multi-species data set. Mol Ecol Resour 13:403–416. https://doi.org/10.1111/1755-0998.12077
Slater GSC, Birney E (2005) Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6:31. https://doi.org/10.1186/1471-2105-6-31
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Stebbins RC (1985) A Field Guide to Western Reptiles and Amphibians. Houghton Mifflin Harcourt, Boston
Supple MA, Shapiro B (2018) Conservation of biodiversity in the genomics era. Genome Biol 19:1–12. https://doi.org/10.1186/s13059-018-1520-3
Swenson NG, Howard DJ (2005) Clustering of contact zones, hybrid zones, and phylogeographic breaks in North America. Am Nat 166:581–591. https://doi.org/10.1086/491688
Thioulouse J, Dray S, Dufour A-B et al (2018) Multivariate analysis of ecological data with ade4. Springer, New York. https://doi.org/10.1007/978-1-4939-8850-1
Vredenburg VT (2004) Reversing introduced species effects: experimental removal of introduced fish leads to rapid recovery of a declining frog. Proc Natl Acad Sci 101:7646–7650. https://doi.org/10.1073/pnas.0402321101
Vredenburg V, Bingham R, Knapp R et al (2007) Concordant molecular and phenotypic data delineate new taxonomy and conservation priorities for the endangered mountain yellow-legged frog. J Zool 271:361–374. https://doi.org/10.1111/j.1469-7998.2006.00258.x
Vredenburg VT, Knapp R, a, Tunstall TS, Briggs CJ, (2010) Dynamics of an emerging disease drive large-scale amphibian population extinctions. Proc Natl Acad Sci U S A 107:9689–9694. https://doi.org/10.1073/pnas.0914111107
Weisrock DW, Hime PM, Nunziata SO et al (2018) Surmounting the large-genome “problem” for genomic data generation in salamanders. In: Hohenlohe PA, Rajora OP (eds) Population genomics: wildlife. Springer, Cham, pp 115–142
Whiteley AR, Fitzpatrick SW, Funk WC, Tallmon DA (2015) Genetic rescue to the rescue. Trends Ecol Evol 30:42–49. https://doi.org/10.1016/j.tree.2014.10.009
Acknowledgements
This work was funded by National Science Foundation LTREB DEB-1557190 and 1556494 and Biology Integration Institute DBI-2120084. Amplicon sequencing done at IBEST Genomics Resources Core at the University of Idaho was supported in part by National Institutes of Health COBRE grant P30GM103324. Exome sequencing was done at the QB3 Genomics Facility, UC Berkeley, Berkeley, CA, RRID:SCR_022304. All sample collections were authorized by research permits provided by NPS, USFWS, CDFW, and the Institutional Animal Care and Use Committees of UC Berkeley, UC Davis, and UC Santa Barbara. This is contribution 851 of the U.S. Geological Survey Amphibian Research and Monitoring Initiative. We thank Beth Shapiro, Dan Rokshar, and Adam Session for providing the whole genome assembly for Rana muscosa. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government. We acknowledge Dr. Rebecca Gooley for providing several tutorials on ROH analyses to AQB, including sharing code, running RZooROH on a preliminary dataset (prior to final sample selection and filtration), creating preliminary figures, and discussing the conservation implications of these results with AQB. We thank Ke Bi for invaluable bioinformatics support. We thank Elizabeth Gallegos, Cynthia Hitchcock, and other USGS staff for field sampling.
Funding
This work was funded by the National Park Service (to RAK), National Science Foundation LTREB DEB-1557190 (to CJB, RAK), LTREB DEB- 1556494 (to EBR), BII DBI-2120084 (to CJB, RKN, EBR), and US Fish and Wildlife.
Author information
Authors and Affiliations
Contributions
A.Q.B. and A.P.R. wrote the main manuscript text. A.P.R., E.B.R., A.Q.B., L.L.S, and R.A.K. designed the study. A.P.R. performed lab work and data cleaning/analysis. A.Q.B. analyzed the data and made the figures. L.L.S. and H.K performed critical lab work. R.A.K., D.M.B, C.J.B., A.R.B., and R.N.F. provided samples and helped write the manuscript. E.B.R. provided funding and helped write the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Byrne, A.Q., Rothstein, A.P., Smith, L.L. et al. Revisiting conservation units for the endangered mountain yellow-legged frog species complex (Rana muscosa, Rana sierrae) using multiple genomic methods. Conserv Genet 25, 591–606 (2024). https://doi.org/10.1007/s10592-023-01568-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-023-01568-5