
Abstract
Bone is established as the preferred site of breast cancer metastasis. However, the precise mechanisms responsible for this preference remain unidentified. In order to improve outcome for patients with advanced breast cancer and skeletal involvement, we need to better understand how this process is initiated and regulated. As bone metastasis cannot be easily studied in patients, researchers have to date mainly relied on in vivo xenograft models. A major limitation of these is that they do not contain a human bone microenvironment, increasingly considered to be an important component of metastases. In order to address this shortcoming, we have developed a novel humanised bone model, where 1 × 105 luciferase-expressing MDA-MB-231 or T47D human breast tumour cells are seeded on viable human subchaodral bone discs in vitro. These discs contain functional osteoclasts 2-weeks after in vitro culture and positive staining for calcine 1-week after culture demonstrating active bone resorption/formation. In vitro inoculation of MDA-MB-231 or T47D cells colonised human bone cores and remained viable for <4 weeks, however, use of matrigel to enhance adhesion or a moving platform to increase diffusion of nutrients provided no additional advantage. Following colonisation by the tumour cells, bone discs pre-seeded with MDA-MB-231 cells were implanted subcutaneously into NOD SCID mice, and tumour growth monitored using in vivo imaging for up to 6 weeks. Tumour growth progressed in human bone discs in 80 % of the animals mimicking the later stages of human bone metastasis. Immunohistochemical and PCR analysis revealed that growing MDA-MB-231 cells in human bone resulted in these cells acquiring a molecular phenotype previously associated with breast cancer bone metastases. MDA-MB-231 cells grown in human bone discs showed increased expression of IL-1B, HRAS and MMP9 and decreased expression of S100A4, whereas, DKK2 and FN1 were unaltered compared with the same cells grown in mammary fat pads of mice not implanted with human bone discs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The development of metastatic disease, in most cases bone metastases, marks the progression of breast cancer to an incurable stage. Median survival after diagnosis of skeletal involvement is around 2 years and there are currently no available therapies that prevent or predict the occurrence of bone metastases [1]. This is in marked contrast to the large improvements in outcome for patients with organ-confined breast cancer seen in the past two decades.
The precise cellular and molecular mechanisms responsible for bone metastases formation remain elusive, and progress in this field is hampered by the lack of human material available for study [2–6]. Sampling of metastatic lesions from bone is rarely done, and then mainly in connection with surgery to stabilise pathological fractures caused by cancer-induced bone erosion. Samples are therefore not only limited in number and tumour content, but the quality is often poor and almost invariably collected from patients that have undergone extensive therapy [7]. As a result, the majority of studies of breast tumour-bone cell interactions are carried out using xenograft models, where human tumour cells are implanted in immunocompromised mice reviewed in ([8–11]). Although useful, these models have major limitations. In particular, they lack the human bone microenvironment that is thought to play a key part in both initiation and progression of bone metastasis, and may also modify the response to therapy [12]. Attempts to improve the relevance of bone metastasis models have been made, mainly through implantation of human bone samples in immunocompromised mice, followed by implantation of human tumour cells in the mammary fat pads in the same animals [13–16]. This establishes a model that mimics all stages of human bone metastasis, including spread from a primary site to bone via the circulation.
The importance of the human bone microenvironment for tumour cell homing is supported by the discovery that the tumour cells do not colonise the mouse skeleton, but preferentially metastasise to the human bone discs [2, 13–16]. The main limitation with this model, however, is the low and highly variable frequency of metastasis to the human bone implant (commonly around 30 %) and the extensive time before metastases are detected (at least 5–6 months). This limits the utility of the model and may explain why few studies have been published since it was first described [17]. In particular, the low frequency of metastasis makes the model unsuitable for investigating effects of therapies, as it is difficult to assess whether a reduction in animals with metastases is the result of the intervention or just reflecting variability in the rates of tumour colonisation of bone. In addition, many researchers do not have ready access to the fresh human bone samples required. When using this model in our laboratory we found that the very low number of bone metastasis available for analyses from each experiment prevented comprehensive screening of molecules involved, as well as assessment of therapeutic effects. We therefore developed a new version of the model, bypassing the escape from the primary tumour and dissemination through the circulation. Here we describe how pre-seeding of human bone discs with human breast cancer cells in vitro, followed by implantation of tumour-cell bearing discs in immunocompromised animals, results in development of tumours in the human bone in the majority of animals over the following 3–5 weeks. By implanting two tumour-bearing bone discs in each animal we were able to dramatically increase the amount of human tumour and bone material available for subsequent analyses.
We have previously demonstrated that during the process of breast cancer bone metastasis, different molecular profiles are associated with homing to compared with colonisation of bone [18]. Characterisation of the bone seeking MDA-MB-231-IV cells made in house via repeated in vivo passaging through bone (described Nutter et al. [18]) demonstrated that bone homing was associated with decreased expression of the cell adhesion molecule fibronectin and the calcium signal binding protein S100A4, as well as increased expression of IL-1B. In contrast, bone colonisation was associated with increased fibronectin expression and upregulation of molecules that influence signal transduction pathways and breakdown of extracellular matrix, including HRAS and MMP9. In the current study we have investigated the expression profile of these molecules in MDA-MB-231 cells growing in human bone in vivo compared with the same cell line growing in mammary fat pads of NOD SCID mice. These data have been used to identify whether the microenvironment in human bone discs can promote changes in breast cancer cells that are associated with metastasis and therefore assess the relevance of our model for studying tumour cell/bone cell interactions.
Materials and methods
Animals
All experiments were carried out in 10-week old female NOD SCID nude mice (Charles River, Kent, UK). Mice were maintained on a 12 h:12 h light/dark cycle with free access to food and water. Experiments were carried out in accordance with local guidelines and with home office approval under project licence 40/3531, University of Sheffield, UK.
Tumour cells
eGFP expressing ER positive T47D and eGFP or Luc2 expressing ER/PR negative MDA-MB-231 human breast cancer cells were cultured in DMEM + 10 % FCS (Gibco®, Invitrogen, Paisley, UK). Prior to seeding on bone discs, tumour cells were labelled for multiphoton analysis by incubation for 15 min with 25 µM of 1,1′-dioctadecyl-′, 3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate (DiD) (Life Technologies, Paisley, UK) for visualisation using multiphoton microscopy.
Patient consent and preparation of human bone discs
All patients provided written, informed consent prior to participation in this study. Human bone samples were collected under HTA licence 12182, Sheffield Musculoskeletal Biobank, University of Sheffield, UK. Trabecular bone cores (0.5 cm3) were prepared from the femoral heads of patients undergoing hip replacement surgery. Briefly, femoral heads were cut into 5 mm slices using an Isomet 4000 Precision saw (Buehler) with Precision diamond wafering blade (Buehler). 5 mm wide discs were cut using a bone trephine before storing in sterile PBS at ambient temperature.
Seeding of human bone discs with tumour cells and implantation in mice
Bone discs were kept in static or moving (oscillating at 50 rpm) cultures in DMEM + 10 % FBS with media changed every 48 h. Discs were seeded on day 2 with 1 × 105 DiD labelled MDA-MB-231-luc2 cells either with or without BD matrigel™ (BD Biosciences, Oxford, UK). Analysis was performed 10 days post seeding. Tumour-cell bearing bone discs were implanted subcutaneously into the left and right flanks of 10-week old, female NOD SCID mice (n = 15) under isofluorane anaesthesia. Mice received an injection of 0.01 ml of an analgesic (0.3 mg/ml? Vetergesic) and Septrin was added to the drinking water for 1 week following bone implantation. Tumour growth in the bone discs was monitored weekly using an IVIS (luminol) system (Caliper Life Sciences) following s.c. injection of 30 mg/kg D luciferin (Invitrogen). 24 h prior to sacrifice mice received an intra-peritoneal injection of 30 mg/kg Calcein (Sigma-Aldrich, Pool, UK) to enable visualisation of newly formed bone. Mice were culled 7, 14, 21 and 28 days post implantation (n = 5 per group) and bone discs and serum collected for downstream analyses.
Mammary fat pad injection of MDA-MB-231 cells
5 × 105 MDA-MB-231 luc2 cells in 10 μl (30 % Matrigel/70 % PBS) were injected into the left and right hind mammary fat pads. Tumour growth was monitored by IVIS imaging and tumours removed 6-weeks after injection. 50 % of mammary tumours were fixed in 4 % paraformaldehyde for histological analysis and 50 % were stored at −80 °C prior to RNA extraction.
Multiphoton microscopy
Human bone discs were removed from the mice, snap frozen in liquid nitrogen and embedded in Cryo-M-Bed embedding compound (Bright Instrument Co. Ltd, Huntingdon, UK) before being trimmed to create a flat surface using a Bright OTF Cryostat with a 3020 microtome (Bright Instrument Co. Ltd, Huntingdon, UK). A Zeiss LSM510 NLO upright multiphoton microscope (Carl Zeiss Microscopy Ltd, Cambridge, UK) was then used to image the entire surface area from 0 to 100 μm in depth. A 633 nm HeNe laser was used to detect DiD labelled cells, whereas calcein and bone were detected using the 900 nm Chameleon multiphoton laser (Coherent, Santa Clara, CA.) The images were subsequently reconstructed with the LSM software 4.2 (Carl Zeiss Microscopy Ltd, Cambridge, UK).
Microcomputed tomography
Analysis of bone volume was carried out using a Skyscan 1172 X-ray–computed microtomograph (Skyscan, Aartselaar, Belgium) equipped with an X-ray tube (voltage, 49 kV; current, 200 mA) and a 0.5-mm aluminum filter. Pixel size was set to 7 µm. For each sample, cross-sectional images were reconstructed with NRecon software (version 1.4.3, Skyscan). Volume of interest was defined on the two-dimensional acquisition images by drawing a 4 mm circle. Trabecular bone volume fraction (Bone volume/tissue volume; BV/TV), the ratio of the volume of bone present (BV) to the volume of the cancellous space (TV), was calculated for 3 mm of the bone. Modeling and analysis were performed with the use of CTAn (version 1.5.0.2) and CTvol (version 1.9.4.1) software (Skyscan).
Measurement of serum marker of bone turnover
Human tartrate-resistant acid phosphatase (TRAP5b), 2 collagen type 1 cross-linked C-telopeptide (CTX) and intact pro collagen type-1N propeptide (P1NP) were measured in tissue culture medium and mouse serum using the IDS-iSYS automated immunoassays (Immunodiagnostic Systems, Boldon, UK).
Histology
Bone discs were fixed in 4 % paraformaldehyde and analysed by uCT prior to decalcification in a solution of 1 % paraformaldehyde/0.5 % EDTA in PBS for 4 weeks with change of solution every 3–4 days and then embedded in paraffin wax. Osteoclasts were identified by tartrate-resistant acid phosphatase (TRAP) staining of 5 μM histological sections. Briefly, dewaxed sections were incubated in acetate–tartrate buffer at 37 °C for 5 min followed by incubation in naphthol AS-BI phosphate, dimethylformamide in acetate-tartrate buffer for 30 min at 37 °C. Sections were placed in a solution containing sodium nitrite, pararosaniline and acetate-tartrate buffer for 15 min at 37 °C, before counterstaining with haematoxylin. Images of the tumour-bearing bones were generated using a ScanScope digital slide scanner and software (Aperio, CA, USA).
Immunohistochemistry
The presence of osteoblasts, infiltrating macrophages, human- and mouse-derived blood vessels was detected by immunohistochemistry using human specific osteocalcin (M184, Takara Bio Inc, Japan), human tumour cells by a COX IV antibody (4850, Cell Signalling), mouse macrophages by F4/80 (MCA497R, Serotech, Kidlington, UK), human specific CD31 (ab76533, Abcam, Cambridge, UK) and mouse specific CD31 (557355, Cambridge Biosciences, Cambridge, UK) antibodies, respectively. To confirm protein expression of molecules associated with tumour growth in bone, histological sections from mammary and bone tumours were incubated with human HRAS (ab97488 1:200, Abcam), human S100A4 (ab40722 1:250, Abcam) and human DKK2 (an38594, 1:100, Abcam). Staining was visualised with corresponding biotin-conjugated secondary antibodies (Vector Laboratories, 1:200) and DAB substrate kit (Vector Laboratories, Peterborough, UK).
Real time PCR
tRNA was extracted with Trizol (Invitrogen AB, Stockholm, Sweden) in combination with RNA clean and concentrator kit (Zymo Research Corporation, Irvine, CA, USA) prior to reverse transcription using High-Capacity RNA-to-cDNA™ Kit (Life technologies-Applied Biosystems). Relative mRNA expression of target genes: DKK2 (Hs00205294_m1), S100A4 (Hs00243202_m1), IL-1B (Hs00174097_m1), Hras (Hs00610483_m1), Fn1 (Hs00365058_m1) and Mmp9 (Hs00234579_m1) were compared with the housekeeping gene GAPDH (Hs01569256_m1) (Applied Biosystems, Warrington, UK) using TaqMan universal master mix (Applied Biosystems) and ABI 7900 PCR system (PerkinElmer, Foster City, CA, USA). Relative mRNA was determined using ΔCT (target gene)− CT (GAPDH). Data were analysed using DataAssist™ Software version 3.01 (Applied Biosystems). To assess gene expression of human MDA-MB-231 breast cancer cells growing in human bone discs, relative mRNA expression of target gene compared with GAPDH were analysed separately in tumour bearing bones and non-tumour bearing bones from the same patient. Gene expression in MDA-MB-231 cells in bone was calculated using the formula ΔCT for tumour in bone − ΔCT non-tumour bearing bone. All Taqman assays used were human specific and no expression was detected in mouse mammary fat pads, therefore, gene expression changes between tumour cells growing in bone and those in the fat pad were calculated using the formula: (ΔCT for tumour in bone − ΔCT non-tumour bearing bone) − ΔCT tumour in mammary fat pad. Genes changed in expression twofold or more with a P value of >0.5 by students t test were considered significant.
Statistical analysis
Statistical analysis for non-PCR experiments was by unpaired T test using GraphPad PRISM® software version 5.0. Statistical analysis of real time PCR experiments was carried out using DataAssist™, version 3.01. P values were calculated by unpaired T test and adjusted by Benjamini-Hochberg false discovery rate test. Statistical significance was defined as P ≥ 0.05.
Results
Bone disc variability between different donors
The quality of the donor bone is of major importance and this varies greatly depending on a number of factors, including age, pathology and therapies used to treat the underlying condition. In our case the age of the donors ranged from 49 to 68, with the majority undergoing hip replacement surgery for osteoarthritis and inflammatory arthritis of the hip. The quality of each bone sample was assessed following slicing of the isolated femoral head, and only samples with a high content of intact bone were used in subsequent experiments. We found that the areas around the rim of the disc near the cortical bone surface (Fig. 1a) consistently contained the highest quality trabecular bone structure (Fig. 1b, c). Despite differences in age, diagnosis and treatment, discs isolated from this area obtained from different donors were found to have comparable bone volume as measured by uCT (Fig. 1d).

Bone disc integrity/volume does not differ significantly between donors. Bone discs (5 mm3) were cut from slices of human femoral heads (a) and analysed by μCT for trabecular structure (b) and 3D reconstruction (c). d is a comparison of bone volume/tissue volume (BV/TV) from three different donors was performed on bone discs (n = 6–7 per donor). Data represent mean ± SEM, NS by one-way ANOVA
In vitro maintenance of bone discs: optimal serum concentration
In order to establish how best to maintain the viability of the different cell types as well as the integrity of the bone discs, we compared the bone volume:tissue volume (BV/TV), number of cells present in the medium and the levels of the resorption marker CTX in bone discs cultures grown in the presence of 0–10 % FCS (Fig. 2). There was no significant difference in the number of cells in the medium between bone discs grown in different serum concentrations after a 48 h incubation period (A), supporting that there was no increased tendency of cells to migrate out of the discs with media containing either a high or low concentration of serum. Bone volume was also unaffected by the serum content of the media (B). Following culture in 10 % serum there was a small but significant increase in the level of the resorption marker CTX serum compared with all of the other concentrations investigated (P = 0.048). There was also a significant increase in CTX in the medium isolated from bones cultured in serum compared with those cultured in serum free medium (CXT concentrations were 0.09 ± 0.02 in serum free medium compared with 0.21 ± 0.03 in 1 % medium (P < 0.05); 0.32 ± 0.14 in 2 % medium (P, 0.05); 0.21 ± 0.05 in 5 % medium (P = 0.05) and 0.70 ± 0.43 in 10 % medium (P < 0.05). This increase in CTX implies that serum is required for activation of osteoclasts and that presence of serum increased viability and bone cell activity, subsequent experiments were therefore carried out in DMEM + 10 % FCS.

Optimal serum concentration for maintaining discs in vitro. Bone discs were cultured in the presence of increasing concentrations of foetal calf serum (FCS). Media and bone discs were harvested at 48 h (n = 4 per concentration) and analysed for effect on the number of cells shed into the media (a). Bone architecture was assessed by measuring volume/tissue volume (BV/TV) (b) and CTX secretion into the media was analysed by ELISA (c). Data represents mean ± SEM
In vitro maintenance of bone discs: static versus moving cultures
We next investigated the effects of extending the in vitro bone disc culture up to 7 days, as this is the time required to establish growing tumour colonies for subsequent in vivo implantation. For these experiments all medium was removed to enable enumeration of cells and exchanged with fresh medium every 24 h. As shown in Fig. 3a, a high number of cells (both dead and viable) appeared in the media during the first 5 days in culture, but by day 6 the bone discs had equilibrated and few cells could be detected in the surrounding medium. Bone volume remained stable over the 7 days (Fig. 3b), demonstrating that there is no loss of bone integrity during short-term cultures. In static cultures, diffusion of nutrients through the bone might be hindered resulting in necrosis, whereas a moving culture system may help to overcome this. We therefore compared bone integrity between discs kept in static and moving cultures in DMEM + 10 % FBS. BV/TV and CTX concentrations (a measure of osteoclast activity) were measured after 12 days in culture (n = 2–3 per group). As shown in Fig. 3c and d, a moving culture system provided no extra benefit to bone disc integrity (as determined by uCT) or viability of osteoclasts (as determined by CTX concentration).

Bone disc integrity over time and static versus moving cultures. Bone discs were cultured in DMEM + 10 % FCS for 1–7 days with fresh media added every 48 h. The total cell count in the media decreased with time (a n = 6 per time point). Bone volume/tissue volume remained unaltered over time (b n = 2–3 per time point). Bone discs were cultured for 12 days under static or moving conditions (n = 2 per group) with fresh media added every 48 h. Movement had no effect on bone volume/tissue volume (c) or CTX secretion (d) compared to static conditions. Data represents mean ± SEM and *P < 0.05 compared with day 1 by students T test
Tumour cells seeded on bone discs: static versus moving cultures
To determine whether the presence of tumour cells affected bone disc integrity in vitro, we measured BV/TV and CTX levels in bone discs 10 days after seeding with 1 × 105 DiD-labelled MDA-MB-231-luc2 cells, with or without matrigel. Matrigel was included to prevent tumour cells from floating off the bone discs, and therefore potentially enhance the rate of bone colonisation. Separate samples were processed for visualisation of the DiD-labelled tumour cells using multiphoton microscopy. As shown in Fig. 4, there was no significant difference in BV/TV between naïve bone and bone discs seeded with tumour cells 10 days after seeding, either in static (P = 0.31 for bones containing tumour cells compared with bone only and P = 0.07 for bones containing tumour cells + matrigel compared with bone only) (Fig. 4a) or moving (P = 0.06 for bones containing tumour cells compared with bone only and P = 0.734 in bone containing tumour cells + matrigel compared with bone only) (Fig. 4b) cultures. Likewise, the concentration of CTX in the medium was not significantly increased in cultures of bone seeded with tumour cells compared to naïve in either culture condition (Fig. 4c, d). In static culture, 0.12 ± 0.01 ng/ml of CTX were detected in the medium from bone only cultures, whereas, addition of MDA-MB-231 cells to the bone resulted in CTX concentrations of 0.31 ± 0.27 ng/ml being secreted into the medium (P = 0.40 compared with control) and 0.30 ± 0.18 ng/ml CTX were secreted into the medium when MDA-MB-231 cells were seeded into bone + matrigel (P = 0.24). In moving culture 0.12 ± 0.02 ng/ml CTX were detected in the medium from bone only cultures whereas 0.17 ± 0.01 ng/ml were detected when MDA-MB-231 cells were cultured in bone (P = 0.07 compared with control) and 0.15 ± 0.06 ng/ml were detected when MDA-MB-231 cells were cultured in matrigel in bone (P = 0.6 compared with control). DiD positive tumour cells were detected in the bone discs from day 2 by multiphoton microscopy in both moving (Fig. 4e) and static (Fig. 4f) cultures, demonstrating successful in vitro colonisation. For subsequent in vivo implantation experiments, tumour cells were seeded on bone discs cultured in the presence of 10 %FCS in static cultures for 7 days.

Tumour cell seeding of bone discs in vitro. 1 × 105 DiD labelled MDA-MB-231-luc2 cells were prepared in either media or 30 % matrigel/media and loaded onto human bone discs 48 h after bone harvest. These were then grown as either static or moving cultures for a further 10 days. The presence of matrigel had no significant effect on bone volume/tissue volume in both static (a) and moving (b) conditions. CTX secretion was also unaffected by the addition of matrigel under both culture conditions (c, d). N = 2–4 per group. Data represents mean ± SEM. e Representative MP images of DiD-labelled MDA231 cells in bone 48 h after seeding
Tumour growth in vitro
Preliminary experiments were carried out using ER-ve MDA-MB-231 cells. In order to determine whether this model could also be used to investigate human tumour cell-bone cell interactions in ER +ve breast cancer cells we compared tumour take and growth of eGFP expressing MDA-MB-231 and ER +ve T47D cells following seeding into human bone discs (Fig. 5): MDA-MB-231 cells grew significantly faster than T47D cells (P < 0.01) and tumour growth was detected by eGFP imaging in 95 % of bones 18 days following seeding with MDA-MB-231 cells compared with 45 % of bones seeded with T47D cells. However, 4 weeks following tumour cell seeding no significant differences in tumour take were observed in human bone discs seeded with MDA-MB-231 or T47D cells implying that both cell types grow equally well when cultured in a human bone environment in vitro.

Tumour growth in bone in vitro. Bone discs were seeded with 1 × 105 MDA-MB-231-GFP or T47D-GFP cells and tumour growth monitored for 28 days. Photomicrographs represent ×40 magnification of MDA-MB-231 and T47D cells growing in bone 28 days after seeding and the graph represents time taken until GFP positive tumour cells are detected in bone
Tumour growth in vivo
Having established the optimal conditions for short-term cultures of the human bone discs, as well as the seeding and subsequent colonization of the discs by tumour cells, we next performed an in vivo study implanting tumour cell-bearing discs in NOD SCID mice. Seeding the bone discs with luciferase-expressing tumours cells in vitro allows monitoring of subsequent colonisation, thereby ensuring that only bone discs with established tumour colonies are implanted into animals. As shown in Fig. 6a, the majority of the bone discs had a strong luciferase signal on the day immediately prior to in vivo implantation, on day 7, indicating initiation of tumour cell growth. Implantation of tumour cell bearing discs into NOD SCID mice resulted in tumour growth in the human bone in 12 out of 15 animals by day 29 (corresponding to 80 % take rate), example images are shown in Fig. 6b.

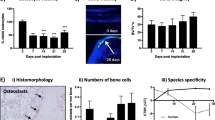
Tumour growth and bone turnover in vivo following implantation of tumour cell-bearing human bone discs. Bone discs were seeded with 1 × 105 DiD labelled MDA-MB-231-luc2 cells in static cultures in vitro and tumour cell colonisation confirmed by luciferase imaging on day 10 (a). Bone discs with confirmed tumour cell colonisation were implanted subcutaneously into the left and right flanks of 10-week old, female NOD SCID mice (n = 15) and tumour growth monitored by in vivo imaging of luciferase up to day 36 (example images from day 29 shown in b). ×40 magnification of histological sections of tumour-bearing human bone sections stained with H&E for identification of osteocytes and osteoblastic cells (c) and TRAP for identification of osteoclasts (d). The different cell types are indicated by arrows. Activity of osteoblastic cells is shown by calcein incorporation in new bone (e) and activity of osteoclasts was measured by ELISA for serum concentrations of human TRAP 5B (f)
Bone cell types present in human bone discs after in vivo implantation
Following the successful generation of human tumours in the human bone discs in vivo, we carried out histological, morphological and biochemical analysis to determine the cellular composition and activity of the bone microenvironment in the tumour bearing human bone discs. As shown in Fig. 6c, d, the main bone cell types (osteoblasts, osteocytes and osteoclasts) were all detected, although the numbers were variable. Empty lacunae indicated some loss of osteocyte viability, probably occurring in the period prior to vascularization of the bone implants. Figure 6E shows new bone formation in human bone discs 24 h after injection of calcein on day 21 and 28, indicating that bone is actively being laid down at these time points. Activity of human osteoclasts in bone discs was detected in the serum of mice by TRAP ELISA (Fig. 6f), however, although we could demonstrate active deposition of new bone human P1NP was below the limit of detection, by ELISA, in mouse blood (data not shown).
Identification of cells in the tumour microenvironment
In order to characterise the human bone tumours generated in this model, we performed an extensive histological analysis of the samples using immunohistochemistry to identify a number of cell types in the tumour microenvironment. Figure 7 shows examples of tumour cells were visualized following staining using an antibody specific for COX4, vessels by antibodies to mouse and human CD31, respectively, and macrophage infiltration using an antibody to F4/80. All these cell types were easily detected in the tumours, supporting that this model system reproducibly generates viable, vascularised, proliferating, human tumours growing in human bone.

Histology of human bone discs after in vivo implantation. ×10 and ×20 magnification of histological sections of tumour-bearing bone discs isolated from animals at 8 and 10 weeks. As indicated, human breast cancer cells were visualized following staining using an antibody specific for COX4, mouse-derived vessels using mouse anti-CD31, human-derived vessels using human anti-CD31, and macrophage infiltration using an antibody to F4/80
Expression profile of molecules associated with bone metastasis
Growth of MDA-MB-231 breast cancer cells in mouse bone have previously been associated with increased expression of interleukin 1B (IL-1B), Harvey rat sarcoma viral oncogene (HRAS) and matrix metalloproteinase 9 (MMP9) and decreased expression of S100 calcium binding protein A4 (S100A4) whereas dickkopf 2 (DKK2) and fibronectin 1 (FN1) are reported to be unaltered compared with the same cells grown in mammary fat pads [18]. We next investigated whether tumour cells growing in the human bone microenvironment also undergo these molecular changes. Real time PCR analysis revealed a significant reduction in S100A4 in MDA-MB-231 cells growing in human bone discs compared with mammary fat (P < 0.01) and significant increases in HRAS (P < 0.001), IL-1B (P < 0.01) and MMP9 (P < 0.001). Expression of DKK2 increased 40.5 ± 18.2 fold but did not reach statistical significance (P = 0.052 by students’ T test) and FN1 gene expression did not change between MDA-MB-231 cells growing in human bone discs or in the mouse mammary fat pad (Fig. 8a). Immunohistochemical staining for DKK2, S100A4 and HRAS confirmed no changes in DKK2, decreased S100A4 and increased HRAS protein in MDA-MB-231 cells growing in human bone discs compared with mouse mammary fat pads (Fig. 8b). These data indicate that our new model of human tumour growth in human bone is suitable for future studies into tumour cell-bone cell interactions.

Differential expression of molecules expressed by MDA-MB-231 cells growing in human bone discs and mouse mammary fat pads. Fold change (±SEM) in gene expression between MDA-MB-231 breast cancer cells grown in human bone discs compared with mouse mammary fat pads (a). ×20 magnification of histological sections of tumors bearing bone discs and tumours in mouse mammary fat pads following immunohistochemical staining for DKK2, S100A4 and HRAS (b). *P < 0.01
Discussion
Current humanised models of cancer metastasis have several limitations that affect their utility and applicability. Low and variable rate of tumour cell colonisation of the human bone as well as lengthy protocols results in poor reproducibility, high costs and time delays. At a mechanistic level, previous models fail to recapitulate the microenvironment of the metastatic site (species-specific responses and/or heterocellular crosstalk) vital for disease progression. Our current protocol aims to satisfy all of these requirements as well as utilising patient bone comparable in age to those likely to be diagnosed with breast cancer bone metastasis for the generation of a clinically relevant model for studying tumour cell-bone interactions.
Development of metastatic disease models relies fundamentally on the ability to recreate the microenvironment of the metastatic site. In the bone it is thought that tumour cells occupy specific niches that are identical to, or overlapping with, the haematopoietic stem cell niche [19, 20]. This niche is made up of adipocytes, fibroblasts and osteoblasts that originate from mesenchymal cells in the marrow and these cells play important roles contributing to the proliferation and differentiation of cancer cells. Once tumour cells begin to proliferate in bone they promote formation of new bone resorbing osteoclasts which leads to a “vicious cycle” in which increased bone resorption releases growth factors from the bone and these in turn promote increased tumour growth [21].
Human bone discs are fully representative of the human bone environment that breast cancer cells metastasise to in a patient population. These discs contain all of the cell types and bone matrix that are involved in breast cancer colonisation and growth in this environment. Many in vitro models have tried to recapitulate these interactions between bone cells and tumour cells including co-culture of breast cancer cells with fibroblasts [22, 23], mesenchymal stromal cells [24] osteoclasts [25], osteoblasts or a mixture of these cell types [26]. When cultured on plastic these models lack the 3D space that is critical in determining cancer cell function (reviewed in [27]). In order to better model the spacial dynamics of human cancer growth in bone tissue engineering technology platforms have been utilised to re-create cell’s naturally occurring environment in order to study cell–cell interactions in vitro. To date the most physiologically relevant version of this model consist of multiple-cell-layered oncogenic constructs built around artificial scaffolds to mimic 3D bone [28]. Although these models have been successfully used to investigate direct interactions between breast cancer cells and other cell types within bone (osteoblasts and mesenchymal cells) [29, 30] they lack faithful anatomical reconstruction of the metastatic site and are unable to mimic the “vicious cycle” between tumour cells and bone cells that is driven by resorption of the bone matrix. Previously, laboratories have used mouse calvaria seeded with breast cancer cells to model the vicious cycle. Although seeding MDA-MB-231 or MCF7 cells onto calvarial bone stimulates osteoclast mediated bone resorption calvarial bone is anatomically very different from subchondral bone found in the femur and this model also lacks human specificity [31]. In breast cancer, bone metastasis is primarily detected in the hip, long bones and vertebra [32]. It is hypothesised that breast cancer cells home to and colonise these sites because they comprise of highly vascularised areas of trabecular bone. This trabecular bone is interspersed with marrow and it is likely that the chemotactic factors contained within the bone marrow are important components of this metastatic process [32]. Calvarial bone lacks this marrow component, possibly accounting for low levels of skull based metastasis observed in cancer patients. Skull based metastasis is reported to occur in around 4 % of cancer patients with the majority being found in patients with late stage breast cancer (between 40 and 55 % of patients with skull metastases) [33, 34]. This condition accounts for ~1 % of all bone metastasis from breast cancer [35] and is primarily found in patients who have previously been diagnosed with disseminated disease in other sites, especially bone, indicating that metastasis to the skull is likely to be a secondary event [36, 37].
We have demonstrated the viability and activity of cells required for this metastatic niche. Furthermore, we have shown that these cells remain in their correct anatomical position within a 3D environment following culture in vitro and in vivo implantation. We are therefore confident that this model will allow future investigations into tumour development based on a number of simultaneous influences rather than those of a single cell type as previously described. Furthermore, bone used in this model is taken from patients who are of a comparable age to those who are most likely to be diagnosed with breast cancer bone metastasis [38] making this a clinically relevant model for studying tumour cell-bone cell interactions that are associated with breast cancer bone metastasis.
Our research group and others have aimed to use humanised models to recapitulate all the different stages of bone metastasis including homing to and colonisation of bone [13–17]. The major disadvantage of such a model has been the poor reproducibility and length of time to secondary tumour development. Whilst our 3D bone disc model forgoes the initial steps of metastasis and focuses on looking at later stages of disease when tumour cells are already seeded in bone, by implanting human bone discs pre-seeded with breast cancer cells into mice we increase tumour take to 80 %. This represents considerable cost and time benefits whilst also addressing the major limitations of the most commonly used in vivo models of bone metastasis; the lack of a human bone microenvironment. The overwhelming literature in this area consists of reports from either xenograft or syngeneic models, in which tumour cells home to and proliferate in murine bone [9–11]. It is likely that there are many important differences between the murine bone microenvironment and human bone colonised by cancer cells, which our model will address.
Study of human bone metastasis is notoriously difficult due to the limited material available [2]. When samples of bone metastases are collected, most often in connection with surgery to repair tumour-induced fractures, these are from late stage disease in patients that have undergone extensive therapeutic intervention [7]. The amount of both bone and tumour in such samples is highly variable, and research to characterise the interactions between bone and tumour cells is lacking. In particular, it is not possible to capture and characterise the very early stages of bone colonisation, an essential step for increasing our understanding of the role played by the bone microenvironment in supporting tumour cell colonisation and subsequent progression. Bone is the main site of metastasis in breast and prostate cancer, with the majority of patients with advanced disease being over the age of 50 and hence with a mature, relatively quiescent skeleton [30]. This means that their bone turnover is low with relatively few sites of active remodelling, providing a completely different bone microenvironment compared to that colonising tumour cells encounter in in vivo model systems. To generate maximum tumour growth in murine bone metastasis (xenograft) models, young animals (typically mice aged <6 weeks) are used [9]. The assumption is that the high bone turnover in mice prior to maturation of the skeleton provides a more supportive soil for tumour cells, compared to that of older animals. Elevating the level of osteoclastic bone resorption in animals with a mature skeleton by OVX or OPG-Fc results in increased tumour take of subsequently injected breast cancer cells, demonstrating the importance of active bone turnover for cancer cell colonisation [10, 39]. This represents a major challenge for humanised bone models, as maintaining bone turnover may not be possible in short term ex vivo models. In the current study low levels of CTX were secreted into the medium from bones cultured for 10 days. These levels did not increase significantly when tumour cells were cultured in the human bone discs. In addition BV/TV remained within the normal range of 20–40 % in bone discs seeded with MDA-MB-231 cells. It is likely that the variability in BV/TV and CTX secreted into the medium from human bone discs in culture make it problematic to identify whether tumour cells are actively resorbing bone in this model, in vitro. However, we do see evidence of osteoclast activity on the surface of bone in contact with tumour cells at later time points (Fig. 6d) when tumour bearing bone has been implanted in vivo. These findings suggest that seeding MDA-MB-231 cells into human bone discs initiates the vicious cycle in which tumour cells stimulate bone resorption leading to the release of growth factors from the bone matrix that can in turn stimulate growth of the tumour cells [21]. Following in vivo implantation but prior to neovascularisation of the human bone, the levels of bone formation and human haematopoiesis is low [17]. In the current study we could only find a very low number of TRAP positive osteoclasts in the human bone discs and the levels of the bone resorption marker TRAP 5b were at the limit of detection. Taken together, these observations explain why bone integrity and volume was stable throughout the incubation period. The low number of osteoclasts was notable, even after 10 weeks of tumour growth in vivo and were only visible in areas of bone directly in contact with tumour, suggesting that prolonged in vivo growth is required to generate substantial bone loss. The differences in remodelling that are required to facilitate growth of human tumour cells in mouse bone (high) compare with human bone (low) may have profound implications for the ways in which tumour cells interact with bone cells in these environments. The human bone disc model provides an ideal recourse for investigating this hypothesis.
The current model represents some of the later stages of bone metastasis progression, after tumour cell colonisation of bone and enables investigations into how tumour cells adapt to grow in a human bone microenvironment. In the current study MDA-MB-231 cells grown in human bone discs showed increased expression of the pro-inflammatory cytokine IL-1B as well as molecules that effect transduction and signalling pathway HRAS and MMP9 and decreased expression of the calcium and signal binding protein S100A4. The cell adhesion molecule DKK2 was increased >40 fold in tumour cells grown in bone compared with the mammary fay pad, however, variation in expression of this molecule between experiments (±18) resulted in this finding not reaching statistical significance. This variation was due to large differences in the expression of DKK2 between different bone discs taken from the same patient (data not shown) suggesting that this molecule may be differentially expressed in different regions of subchondral bone or be correlated with bone turnover. However, It remains inconclusive whether change in DKK2 expression is specifically altered in tumour cells growing in a bone environment and therefore these data warrant further investigation. As well as DKK2, FN1 was also unaltered compared with the same cells grown in mammary fat pads of mice not implanted with human bone discs. We and others have previously shown that the expression profile of these molecules are altered in the same way MDA-MB-231 cells that have homed to and grown in mouse bone following injection into the blood stream [18, 40, 41]. These findings strongly suggest that the microenvironment within human bone discs following in vivo implantation is sufficiently preserved to induce molecular changes in human breast cancer cells that facilitate their growth in this environment.
It is commonly hypothesised that breast cancers can undergo secondary metastasis from bone to other organs. We therefore investigated whether MDA-MB-231 cells could metastasise from bone implants to other organs. Luciferase imaging did not show tumour growth in any organ other than human bones up to and including 10-weeks after implantation implying that tumour cells did not metastasise from the bone implants in this instance. Considering that these cells take 10 weeks or more to metastasise from the mammary fat pad to bone [17], It is highly possible that any tumour cells that have spread from the bone to other sites are not detectable at this time point and may take longer to develop. Further experiments need to be carried out to investigate secondary metastasis from human bone implants at later time points.
In conclusion, we have generated a novel 3D model in which bone turnover is active and all major bone cells are present. This model gives a high tumour take rate, can be used for long or short term studies and has great potential to facilitate research into how tumour cells interact with human bone cells in development of bone metastasis. Importantly it provides the opportunity to establish the mechanisms of early stages of tumour growth in bone, when low numbers of tumour cells are present.
References
Major PP, Cook RJ, Lipton A, Smith MR, Terpos E, Coleman RE (2009) Natural history of malignant bone disease in breast cancer and the use of cumulative mean functions to measure skeletal morbidity. BMC Cancer 9:272
Xia TS, Wang J, Yin H, Ding Q, Zhang YF, Yang HW, Liu XA, Dong M, Du Q, Ling LJ, Zha XM, Fu W, Wang S (2010) Human tissue-specific microenvironment: an essential requirement for mouse models of breast cancer. Oncol Rep 24(1):203–211
Quail DF, Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19(11):1423–1437
Hanahan D, Coussens LM (2012) Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21(3):309–322
Buenrostro D, Park SI, Sterling JA (2014) Dissecting the role of bone marrow stromal cells on bone metastases. Biomed Res Int 2014:875305. Epub 2014 Jun 26
Esposito M, Kang Y (2014) Targeting tumor-stromal interactions in bone metastasis. Pharmacol Ther 141(2):222–233
Cawthorn TR, Amir E, Broom R, Freedman O, Gianfelice D, Barth D, Wang D, Holen I, Done SJ, Clemons M (2009) Mechanisms and pathways of bone metastasis: challenges and pitfalls of performing molecular research on patient samples. Clin Exp Metastasis 26(8):935–943
Rosol TJ, Tannehill-Gregg SH, Corn S, Schneider A, McCauley LK (2004) Animal models of bone metastasis. Cancer Treat Res 118:47–81
Ottewell PD, Woodward JK, Lefley DV, Evans CA, Coleman RE, Holen I (2009) Anticancer mechanisms of doxorubicin and zoledronic acid in breast cancer tumor growth in bone. Mol Cancer Ther 8(10):2821–2832
Ottewell PD, Wang N, Brown HK, Reeves KJ, Fowles CA, Croucher PI, Eaton CL, Holen I (2014) Zoledronic acid has differential antitumor activity in the pre- and postmenopausal bone microenvironment in vivo. Clin Cancer Res 20(11):2922–2932
Brown HK, Ottewell PD, Evans CA, Holen I (2012) Location matters: osteoblast and osteoclast distribution is modified by the presence and proximity to breast cancer cells in vivo. Clin Exp Metastasis 29(8):927–938
Coleman RE, Gregory W, Marshall H, Wilson C, Holen I (2013) The metastatic microenvironment of breast cancer: clinical implications. Breast 22:S50–S56
Kuperwasser C, Dessain S, Bierbaum BE, Garnet D, Sperandio K, Gauvin GP, Naber SP, Weinberg RA, Rosenblatt M (2005) A mouse model of human breast cancer metastasis to human bone. Cancer Res 65(14):6130–6138
Lam P, Yang W, Amemiya Y, Kahn H, Yee A, Holloway C, Seth A (2009) A human bone NOD SCID mouse model to distinguish metastatic potential in primary breast cancers. Cancer Biol Ther 8(11):1010–1017
Holzapfel BM, Thibaudeau L, Hesami P, Taubenberger A, Holzapfel NP, Mayer-Wagner S, Power C, Clements J, Russell P, Hutmacher DW (2013) Humanised xenograft models of bone metastasis revisited: novel insights into species-specific mechanisms of cancer cell osteotropism. Cancer Metastasis Rev 32(1–2):129–145
Xia TS, Wang GZ, Ding Q, Liu XA, Zhou WB, Zhang YF, Zha XM, Du Q, Ni XJ, Wang J, Miao SY, Wang S (2012) Bone metastasis in a novel breast cancer mouse model containing human breast and human bone. Breast Cancer Res Treat 132(2):471–486
Nutter F, Wilkinson JM, Holen I, Ottewell PD (2013) Optomisation of a human specific model of breast cancer bone metastasis. In: 13th International Conference on Cancer-Induced Bone Disease. Florida, USA. Bonekey
Nutter F, Holen I, Brown HK, Cross SS, Evans CA, Walker M, Coleman RE, Westbrook JA, Selby PJ, Brown JE, Ottewell PD (2014) Different molecular profiles are associated with breast cancer cell homing compared with colonisation of bone: evidence using a novel bone-seeking cell line. Endocr Relat Cancer 21(2):327–341
Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verástegui E, Zlotnik A (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410(6824):50–56
Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK (2002) Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res 62(6):1832–1837
Guise TA, Mohammad KS, Clines G, Stebbins EG, Wong DH, Higgins LS, Vessella R, Corey E, Padalecki S, Suva L, Chirgwin JM (2006) Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin Cancer Res 12(20 Pt 2):6213s–6216s
Heneweer M, Muusse M, Dingemans M, de Jong PC, van den Berg M, Sanderson JT (2005) Co-culture of primary human mammary fibroblasts and MCF-7 cells as an in vitro breast cancer model. Toxicol Sci 83(2):257–263
Arrigoni C, De Luca P, Gilardi M, Previdi S, Broggini M, Moretti M (2014) Direct but not indirect co-culture with osteogenically differentiated human bone marrow stromal cells increases RANKL/OPG ratio in human breast cancer cells generating bone metastases. Mol Cancer 13:238
Pasanen I, Pietilä M, Lehtonen S, Lehtilahti E, Hakkarainen T, Blanco Sequeiros R, Lehenkari P, Kuvaja P (2014) Mesenchymal stromal cells from female donors enhance breast cancer cell proliferation in vitro. Oncology 88(4):214–225
Liverani C, Mercatali L, Spadazzi C, La Manna F, De Vita A, Riva N, Calpona S, Ricci M, Bongiovanni A, Gunelli E, Zanoni M, Fabbri F, Zoli W, Amadori D, Ibrahim T (2014) CSF-1 blockade impairs breast cancer osteoclastogenic potential in co-culture systems. Bone 66:214–222
Krawetz R, Wu YE, Rancourt DE, Matyas J (2009) Osteoblasts suppress high bone turnover caused by osteolytic breast cancer in-vitro. Exp Cell Res 315(14):2333–2342
Chong Seow Khoon M (2015) Experimental models of bone metastasis: opportunities for the study of cancer dormancy. Adv Drug Deliv Rev. doi:10.1016/j.addr.2014.12.007
Mastro AM, Vogler EA (2009) A three-dimensional osteogenic tissue model for the study of metastatic tumor cell interactions with bone. Cancer Res 69(10):4097–4100
Krishnan V, Shuman LA, Sosnoski DM, Dhurjati R, Vogler EA, Mastro AM (2011) Dynamic interaction between breast cancer cells and osteoblastic tissue: comparison of two- and three-dimensional cultures. J Cell Physiol 226(8):2150–2158
Marlow R, Honeth G, Lombardi S, Cariati M, Hessey S, Pipili A, Mariotti V, Buchupalli B, Foster K, Bonnet D, Grigoriadis A, Rameshwar P, Purushotham A, Tutt A, Dontu G (2013) A novel model of dormancy for bone metastatic breast cancer cells. Cancer Res 73(23):6886–6899
Curtin P, Youm H, Salih E (2012) Three-dimensional cancer-bone metastasis model using ex vivo co-cultures of live calvarial bones and cancer cells. Biomaterials 33(4):1065–1078
Mastro AM, Gay CV, Welch DR (2003) The skeleton as a unique environment for breast cancer cells. Clin Exp Metastasis 20(3):275–284
Laigle-Donadey F, Taillibert S, Martin-Duverneuil N, Hildebrand J, Delattre JY (2005) Skull-base metastases. J Neurooncol 75(1):63–69
Mitsuya K, Nakasu Y, Horiguchi S, Harada H, Nishimura T, Yuen S, Asakura K, Endo M (2011) Metastatic skull tumors: MRI features and a new conventional classification. J Neurooncol 104(1):239–245
Laigle-Donadey F, Taillibert S, Martin-Duverneuil N, Hildebrand J, Delattre JY (2005) Skull-base metastases. J Neurooncol 75(1):63–69
Greenberg HS, Deck MD, Vikram B, Chu FC, Posner JB (1981 May) Metastasis to the base of the skull: clinical findings in 43 patients. Neurology 31(5):530–537
Stark AM, Eichmann T, Mehdorn HM (2003) Skull metastases: clinical features, differential diagnosis, and review of the literature. Surg Neurol 60(3):219–225
Williams GR, Jones E, MD, Muss HB Treatment of metastatic breast cancer in women aged 65 years and older. Womens Health. http://www.medscape.org/viewarticle/766642
Ottewell PD, Wang N, Brown HK, Fowles CA, Croucher PI, Eaton CL, Holen I (2015) OPG-Fc inhibits ovariectomy-induced growth of disseminated breast cancer cells in bone. Int J Cancer. (Epub ahead of print)
Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J (2003) A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3:537–549
Bellahcene A, Bachelier R, Detry C, Lidereau R, Clezardin P, Castronovo V (2007) Transcriptome analysis reveals an osteoblast-like phenotype for human osteotropic breast cancer cells. Breast Cancer Res Treat 101:135–148
Acknowledgments
This study was supported by a project grant from Weston Park Hospital Cancer Charity, Sheffield, UK. The IVIS imaging system was provided by a grant from Yorkshire Cancer Research, UK. We thank Ms Sophia Sutherland and Dr. Fatma Gossiel for technical assistance and Dr. Paul Heath, University of Sheffield, for his help with genetic analysis. We gratefully acknowledge authorisation to perform the in vivo component of the study under Home Office Project License approval PPL 40/3531 holder Prof. NJ Brown, University of Sheffield, UK.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Holen, I., Nutter, F., Wilkinson, J.M. et al. Human breast cancer bone metastasis in vitro and in vivo: a novel 3D model system for studies of tumour cell-bone cell interactions. Clin Exp Metastasis 32, 689–702 (2015). https://doi.org/10.1007/s10585-015-9737-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-015-9737-y