
Abstract
Calcium (Ca2+) ions are prominent cell signaling regulators that carry information for a variety of cellular processes and are critical for neuronal survival and function. Furthermore, Ca2+ acts as a prominent second messenger that modulates divergent intracellular cascades in the nerve cells. Therefore, nerve cells have developed intricate Ca2+ signaling pathways to couple the Ca2+ signal to their biochemical machinery. Notably, intracellular Ca2+ homeostasis greatly relies on the rapid redistribution of Ca2+ ions into the diverse subcellular organelles which serve as Ca2+ stores, including the endoplasmic reticulum (ER). It is well established that Ca2+ released into the neuronal cytoplasm is pumped back into the ER by the sarco-/ER Ca2+ ATPase 2 (SERCA2), a P-type ion-motive ATPase that resides on the ER membrane. Even though the SERCA2 is constitutively expressed in nerve cells, its precise role in brain physiology and pathophysiology is not well-characterized. Intriguingly, SERCA2-dependent Ca2+ dysregulation has been implicated in several disorders that affect cognitive function, including Darier’s disease, schizophrenia, Alzheimer’s disease, and cerebral ischemia. The current review summarizes knowledge on the expression pattern of the different SERCA2 isoforms in the nervous system, and further discusses evidence of SERCA2 dysregulation in various neuropsychiatric disorders. To the best of our knowledge, this is the first literature review that specifically highlights the critical role of the SERCA2 in the brain. Advancing knowledge on the role of SERCA2 in maintaining neuronal Ca2+ homeostasis may ultimately lead to the development of safer and more effective pharmacotherapies to combat debilitating neuropsychiatric disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Calcium (Ca2+) ions are prominent cell signaling regulators as they carry information for a wide range of cellular processes, from egg fertilization and cell fate to gene expression and development (Berridge et al. 2000; Brini et al. 2014; Orrenius et al. 2003). Notably, Ca2+ is critical for neuronal survival and function. Specifically, Ca2+ ions regulate neuronal and synaptic activity by modulating presynaptic and postsynaptic events, such as neurotransmitter release and dendritic spine density (Berridge et al. 2003; Brini et al. 2014; Carafoli 2003; Lyons and West 2011; Neher and Sakaba 2008; Zucker 1999). It is well established that Ca2+ concentrations in neurons can impact important neurobiological processes such as learning, memory, long-term potentiation/depression (LTP/LTD), and motor function (Artola and Singer 1993; Baker et al. 2008; Cassidy et al. 2013; Kawamoto et al. 2012; Mulkey and Malenka 1992; Salinska et al. 2001; Simonyi et al. 2005). Most importantly, disruption of normal Ca2+ cycling in the brain has been associated with severe neuropsychiatric disorders, including Alzheimer’s disease, Parkinson’s disease, dementia, bipolar disorder, schizophrenia, autism spectrum disorders, and intellectual disabilities (Bezprozvanny and Mattson 2008; Dahl 2017; Earls et al. 2010; Green et al. 2008; Jacobsen et al. 1999; LaFerla 2002).
Nerve cells have developed intricate Ca2+ signaling pathways to couple the Ca2+ signal to their biochemical machinery. The Ca2+ signaling toolkit of the brain consists of ion channels, exchangers, and pumps, located both in the cell membrane and the membranes of intracellular organelles. These effectors, together with Ca2+-binding proteins, G-protein coupled receptors (GPCRs), and transcriptional networks, regulate all of the brain’s Ca2+-related processes (Berridge et al. 1998, 2003; Brini et al. 2014; Fucile 2004; Grienberger and Konnerth 2012; Schwaller 2010). Notably, intracellular Ca2+ homeostasis greatly relies on the rapid redistribution of Ca2+ ions into the diverse subcellular organelles which serve as Ca2+ stores, including the ER. It is well established that Ca2+ released into the neuronal cytoplasm is pumped back into the ER by the sarco-/endoplasmic reticulum Ca2+ ATPase 2 (SERCA2) (Brini and Carafoli 2009; Burdakov et al. 2005; Burk et al. 1989; Camacho and Lechleiter 1993; Clapham 1995; Higgins et al. 2006; Wuytack et al. 2002).
The role of the SERCA2 in the cardiovascular system has been well-characterized owing to its involvement in the regulation of cardiac contractility (Kranias and Hajjar 2012). Even though the SERCA2 is constitutively expressed in the nerve cells, its precise role in brain physiology and pathophysiology is elusive. In the current review we summarize current knowledge on the expression pattern of the different SERCA2 isoforms in the nervous system, and further discuss evidence of SERCA2 dysregulation in various neuropsychiatric disorders.
The Main Components of the Neuronal Ca2+-Handling Toolkit
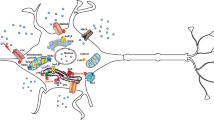
The systems of Ca2+ homeostasis in nerve cells mainly involve Ca2+ buffer proteins that serve as sensors, and a variety of Ca2+-binding transmembrane channels (Berridge et al. 1998, 2000; Brini et al. 2014; Grienberger and Konnerth 2012). Due to its critical role in serving as carrier of critical information, cells must maintain low intracellular Ca2+ levels, so that its concentration can be significantly altered without wasting valuable energy. Interestingly, cytosolic Ca2+ concentrations in resting neurons are approximately 50–100 nM, while Ca2+ levels in firing neurons may increase 10–1000 times (Berridge et al. 2000). Figure 1 depicts all the key players involved in maintaining Ca2+ homeostasis in nerve cells; Ca2+ influx from the extracellular fluid (ECF) is regulated by voltage-gated Ca2+ channels (VGCs), nicotinic acetylcholine receptors (nAchR), ionotropic glutamate receptors (i.e., NMDA-R and AMPA-R), and transient receptor potential (TRP) type C channels. Notably, the ER is a major intracellular Ca2+ storage and mitochondria act as Ca2+ buffers. The SERCA pump and the mitochondrial uniporter facilitate the transport of Ca2+ across the ER and mitochondrial membranes, respectively. Moreover, Ca2+ ions are pumped from internal stores back to the cytosol by the inositol trisphosphate receptors (IP3R) and ryanodine receptors (RyR) that reside on the ER membrane, as well as by the mitochondrial Na+/Ca2+ exchanger (NCX). Finally, the efflux of Ca2+ from the cytosol to the ECF is mediated by the plasma membrane Ca2+ ATPase (PMCA) and the Na+/Ca2+ exchanger (NCX) that reside on the plasma membrane (Berridge et al. 1998; Brini et al. 2014; Duchen 1999; Guerini 1998). Apart from transmembrane channels and Ca2+ buffer proteins, the interaction between intracellular organelles is critical for Ca2+ signaling. Indeed, mitochondria-associated ER membranes (MAM) that physically connect the ER and the mitochondria play an important role in the exchange of inter-organelle Ca2+ signals that regulate cell survival and apoptosis (Bononi et al. 2013; Csordás et al. 2006, 2010; Hayashi et al. 2009; van Vliet et al. 2014).

Neuronal Ca2+-handling: Ca2+ influx in neurons is mediated by calcium-permeable AMPA and NMDA glutamate receptors, nicotinic acetylcholine receptors (nAChR), transient receptor potential type C (TRPC) channels, and voltage-gated calcium channels (VGCC). Ca2+ ions enter into the intracellular Ca2+ stores (i.e., mitochondria and the ER) by the mitochondrial uniporter and the sarco-/endoplasmic reticulum calcium ATPase (SERCA). Ca2+ release from internal stores is mediated by inositol trisphosphate receptors (IP3R) and ryanodine receptors (RyR) that reside on the ER membrane. Ca2+ efflux to the extracellular fluid is mediated by the sodium–calcium exchanger (NCX) and the plasma membrane calcium ATPase (PMCA). Ca2+-binding proteins serve as Ca2+ ion sensors, buffering the cytosolic levels of Ca2+; arrows show the direction of Ca2+ ion movement
Interestingly, Ca2+ storage is one of the functions most commonly attributed to the smooth ER in mammalian cells. When Ca2+ ions are needed, specialized ER channels release Ca2+ from the lumen of the ER to the cytosol. IP3Rs are activated by IP3 produced by phospholipase C (PLC) upon G-protein–coupled receptor (GPCR) activation on the neuronal membrane. On the other hand, RyRs are involved in Ca2+-induced Ca2+-release (CICR). This process occurs when an increase in cytosolic Ca2+ triggers RyRs to release more Ca2+ ions into the cytosol. In addition to Ca2+ concentration in the cytosol affecting RyRs, the intracellular concentration of Ca2+ can also have a stimulatory or inhibitory effect on IP3Rs depending on the concentration of Ca2+ ions (Taylor and Tovey 2010). Specifically, soon after the first IP3-evoked Ca2+ release, exposure of IP3Rs to lower levels of Ca2+ further enhances their response to IP3, whereas higher Ca2+ concentrations further inhibit Ca2+ release. When the concentration of Ca2+ in the cytosol needs to be reduced, Ca2+ may either be transported out of the neuron by means of the PCMA and the NCX, or pumped back into the ER to store for later use. The Ca2+ uptake into the ER lumen is specifically facilitated by the SERCA pump, which resides on the ER membrane.
SERCA2: Structure and Function
SERCAs are P-type ion-motive ATPases and transport two Ca2+ ions from the cytoplasm of cells to the ER lumen per ATP molecule hydrolyzed (Brini and Carafoli 2009; Hasselbach and Makinose 1961; Lee et al. 2002). The different SERCA isoforms comprised a single polypeptide chain, 1000 amino acids in length, and 110 kDa in weight (Dally et al. 2006; MacLennan 1970; MacLennan et al. 1985; Toyoshima and Inesi 2004). Post-translationally, the folded protein resides on the ER membrane, with its ten transmembrane α-helices, short luminal loops, and three cytosolic domains (Fig. 2). These ten transmembrane domains are critical for the function of the protein. Specifically, transmembrane domains M2, M5, M6, and M8 form the SERCA Ca2+ channel, whereas transmembrane domains M4–M6 facilitate in Ca2+ transportation across the ER membrane (Guerini 1998; Zhang et al. 1998). Moreover, four of the transmembrane α-helices (M2–M5) extend beyond the ER membrane and protrude into the cytosol, forming three cytosolic domains (Carafoli and Brini 2000; Toyoshima and Inesi 2004); these domains (A, P, and N) are separated from the transmembrane domains, by a stalk sector. The A domain, or the actuator domain, is created by the cytosolic extension of the M2 and M3 domains; it includes the N-terminus, the Lys120, and Thr245, critical for the binding and release of Ca2+ ions. The P (phosphorylation) and N (nucleotide-binding/hinge) domains are located between the M4 and M5 cytosolic loop. Specifically, the P domain contains the Asp351 residue on which the γ-phosphate binds, forming the high-energy phosphorylation intermediate during the phosphorylation reaction cycle, whereas the N domain includes the nucleotide and ATP-binding sites, with three residues (Lys515, Lys492, and Lys684) playing an important role in the binding process (Brini et al. 2017; Carafoli et al. 2001; Møller et al. 2005; Periasamy and Kalyanasundaram 2007; Toyofuku et al. 1992; Toyoshima and Inesi 2004).

SERCA2 structure: SERCA2 is a P-type Ca2+ ATPase that resides on the SR/ER membrane, protruding into the cytosol. It consists of 10 transmembrane helices (M1–10), a cytosolic stalk domain, and three main domains, A, P, and N. The A domain is the actuator domain, the N domain is responsible for nucleotide-binding, and the P domain accounts for the phosphorylation domain of the enzyme. All SERCA2 isoforms present a very well-conserved structure, but differ in the length of the C-terminal, with SERCA2b isoform having the most extended carboxyl terminal, potentially forming an eleventh transmembrane domain (M11)
Despite the plethora of SERCA isoforms, the protein structure is highly conserved, as all proteins are derived by tissue-dependent alternative splicing of three genes; SERCA1-3 (or ATP2A1-A3 in humans), with distinct expression patterns (Brandl et al. 1986; Gunteski-Hamblin et al. 1988; Lytton and MacLennan 1988; MacLennan et al. 1985). The derived protein isoforms show many similarities but differ in the length of their C-termini (Fig. 3) (Gunteski-Hamblin et al. 1988; Korczak et al. 1988; Zarain-Herzberg et al. 1990). The alternative splicing of the SERCA1 gene results to the formation of SERCA1a and SERCA1b isoforms that are selectively expressed in mature fast-twitch muscle fibers and neonatal skeletal muscle fibers, respectively (Brandl et al. 1987, 1986). Additionally, four SERCA2 splice variants (SERCA2a-d) have been currently identified with high similarities in the 5′-end, but different C-termini (Brandl et al. 1986; Dally et al. 2006, 2009; Gelebart et al. 2003; Gunteski-Hamblin et al. 1988; Lytton et al. 1989; Zarain-Herzberg et al. 1990). Specifically, SERCA2a (997 aa; 110 kDa), a cardiac and slow-twitch muscle-specific protein isoform, has a short C-terminus that consists of 4 aa (NYLEP/AILE), whereas the SERCA2b isoform (1042 aa; 115 kDa) which is found in smooth muscle and non-muscle tissues, has a longer C-terminus of 49 aa (NYLEP/GKEC-4laa-MFWS) (Gunteski-Hamblin et al. 1988; Lytton and MacLennan 1988). This extended C-terminus is believed to penetrate the ER membrane, creating an eleventh transmembrane α-helix (also known as 2b-tail), altering the function of this isoform (Lytton et al. 1992; Verboomen et al. 1992, 1994). The notion for the formation of the 2b-tail is supported by immunohistochemical evidence showing that the SERCA2a and SERCA2b C-termini lie on opposite sides of the ER membrane; the SERCA2a C-terminus extends into the cytosol, whereas the SERCA2b C-terminus protrudes into the ER lumen (Campbell et al. 1992). The SERCA2c isoform, has been recently identified and is believed to be expressed in monocytes and cardiac tissue. This isoform derives from the inclusion of a short coding sequence in intron 20, including an in-frame stop codon (Dally et al. 2006, 2009; Gelebart et al. 2003). Its size is similar to the SERCA2a (999 aa, 110 kDa), while its C-terminal sequence is longer than SERCA2a by 2 amino acids (NYLEP/VLSSEL) (Dally et al. 2009). A fourth SERCA2 mRNA variant, SERCA2d, has also been characterized in skeletal muscle, but its protein isoform is yet to be identified (Kimura et al. 2005). Similar to the other SERCA isoforms, the alternative splicing of the SERCA3 gene results in six different isoforms (SERCA3a-f), differing at least 36 aa residues from each other (Bobe et al. 1998; Poch et al. 1998).

The primary structure of the carboxyl termini of the SERCA2a-d isoforms: The structure of the SERCA2 isoforms is highly conserved but their carboxyl termini differ (3′-end). Slashes mark the splice sites
Despite the variety of protein isoforms, it is well established that SERCAs serve to pump Ca2+ ions from the cytosol into the ER lumen, a process that reduces the cytosolic Ca2+ concentrations, and replenishes ER Ca2+ stores. Throughout a cycle of conformational alternations between a high-Ca2+-affinity (E1) state and a low-Ca2+-affinity (E2) state, two Ca2+ ions cross the ER membrane from the cytosol into the ER lumen, against their concentration gradient and at the expense of ATP (Brini and Carafoli 2009; Dode et al. 2003; Hao et al. 1994; Hasselbach and Makinose 1961; Lee et al. 2002; Periasamy and Kalyanasundaram 2007; Salvador et al. 1998; Vandecaetsbeek et al. 2009; Yu et al. 1993). In its native E1 state, SERCA binds Ca2+ ions on its cytoplasmic high-affinity sites. Once the Ca2+-binding sites are occupied, ATP-binding and hydrolysis are triggered, phosphorylating SERCA. The phosphorylation of the enzyme subsequently alters the conformation of the transmembrane α-helices (E2 state) leading to the release of the two Ca2+ ions in the ER lumen. Once the Ca2+ ion transport is completed, the pump is dephosphorylated, and returns to the E1 state (Carafoli and Brini 2000; Møller et al. 2005; Olesen et al. 2004; Periasamy and Kalyanasundaram 2007; Vandecaetsbeek et al. 2009). SERCA’s apparent affinity for Ca2+ ions may be affected by several factors, including alterations in cellular Ca2+ ion concentrations and pH, as well as internal mutations in the SERCA genes (Lee et al. 2002; Periasamy and Kalyanasundaram 2007; Vandecaetsbeek et al. 2009; Yu et al. 1993). Notably, functional studies have indicated that SERCA1a and SERCA2a isoforms show similar affinities for Ca2+, as well as similar catalytic turnover rates, while SERCA2b presents two times the affinity for Ca2+ and half the turnover rate, as compared to the SERCA2a isoform (Lytton et al. 1992; Verboomen et al. 1994). It is thought that SERCA2c could perform in a local Ca2+-rich environment because SERCA2c presents the lowest affinity for Ca2+ out of the three SERCA2 isoforms (Dally et al. 2006). Last but not least, the different SERCA3 isoforms have similar affinities for Ca2+ (Bobe et al. 2004; Martin et al. 2002). However, SERCA3a presents a similar turnover rate when compared to the SERCA2b isoform, whereas both SERCA3b and SERCA3c possess a higher turnover rate. However, all SERCA3 isoforms have a lower affinity for Ca2+ compared to SERCA2b (Dode et al. 1998).
SERCA2 Expression Pattern in the CNS
Currently, the expression of at least ten distinct SERCA isoforms has been identified in mammalian cells (Baba-Aissa et al. 1998; Periasamy and Kalyanasundaram 2007). As discussed above, the vertebrate SERCA isoforms are encoded by alternatively spliced transcripts of the SERCA1-3 genes (Brandl et al. 1986; Gunteski-Hamblin et al. 1988; Lytton and MacLennan 1988; MacLennan et al. 1985). Despite the differences in these three genes, all of them have been largely conserved with none being more than 30% different than the others (Periasamy and Kalyanasundaram 2007). Notably, the SERCA2 is the isoform that is predominately expressed in the CNS (Gunteski-Hamblin et al. 1988). In fact, SERCA2 mRNA expression has been detected with in situ hybridization in Purkinje neurons of the cerebellum, followed by expression in the thalamus, the cortex, the pontine nuclei, and the mitral cells of the olfactory bulbs (Miller et al. 1991). Additionally, subsequent immunoblotting studies in the pig cerebellum have confirmed that SERCA2 resides in the Purkinje cells, the granule cells, and the cerebellar glomeruli (Sepulveda et al. 2004), while SERCA1 and SERCA3 expression is confined to the cerebellar Purkinje neurons (Baba-Aissa et al. 1996; Wu et al. 1995).
Interestingly, the expression pattern of the three SERCA2 isoforms in excitable tissues is quite divergent. SERCA2a is strongly expressed in cardiac and slow skeletal muscle fibers, while it is moderately expressed in smooth muscle cells; SERCA2a expression in the brain is weak and confined in the cerebellar Purkinje neurons and the granular cell layer, as well as in the giant cells of the reticular formation in the brainstem (Baba-Aissa et al. 1996; Campbell et al. 1993; Plessers et al. 1991). On the other hand, SERCA2b is ubiquitously expressed in all cell types, including neurons, cardiac muscle fibers, slow skeletal muscle fibers, and smooth muscle cells (Gunteski-Hamblin et al. 1988; Lytton and MacLennan 1988). Furthermore, SERCA2b is the only SERCA isoform expressed in astrocytes, as shown by recent immunoblotting data using astrocytes isolated from the rat cerebral cortex (Morita and Kudo 2010). Subsequently, the universal expression of the SERCA2b in mammalian cells has led to the consideration of this SERCA isoform as an ER housekeeping protein (Burk et al. 1989; Lytton and MacLennan 1988; Lytton et al. 1989). Last but not least, recent immunoblotting data suggest that the SERCA2c isoform is also expressed at low levels in the brain, but it is more widely expressed in epithelial, mesenchymal, and hematopoietic cells (Dally et al. 2006, 2010; Gelebart et al. 2003).
Remarkably, earlier immunoblotting and sequence analysis studies have identified ubiquitous SERCA2b mRNA and protein expression in both the cerebrum and the cerebellum of the vertebrate brain (Burk et al. 1989; Miller et al. 1991; Plessers et al. 1991). Indeed, immunohistochemical and functional studies have confirmed the global expression of SERCA2b protein in the vertebrate brain (Baba-Aissa et al. 1996; Campbell et al. 1993; Salvador et al. 2001; Sepulveda et al. 2004). Interestingly, Campbell et al. (1993) reported that SERCA2a and SERCA2b are co-expressed in both cerebellar Purkinje cells and cerebral nuclei, but in different ratios, leading to the hypothesis that different brain regions have specific requirements for each of the two SERCA2 isoforms (Campbell et al. 1993). Further immunoblotting and in situ hybridization studies conducted by Baba-Aissa et al. (1996) revealed that the highest levels of SERCA2b were expressed in the Purkinje neurons, followed by the hippocampal pyramidal cells and the cerebral cortical layers II–V. More recent studies by Salvador et al. (2001) further confirmed the universal expression of SERCA2b in subcellular fractions (i.e., microsomes, synaptosomes, and synaptic plasma membrane vesicles) derived from the pig brain. Indeed, SERCA2b expression was identified in all three fractions, but no other isoform was expressed whatsoever. Moreover, the distribution of SERCA2b isoform differed among the fractions, with the microsomes having the highest concentration of SERCA2b, followed by the synaptosomes and the synaptic plasma membrane vesicles (Salvador et al. 2001). Further studies confirmed the universal expression of SERCA2b in the pig cerebellum, with the highest levels found in the soma, the trunk, and the proximal dendritic branches of Purkinje neurons, as well as in the glomeruli of the cerebellar granule layer (Sepulveda et al. 2004). Notably, the weakest expression of SERCA2b was detected in the hypothalamus and the substantia nigra. (Baba-Aissa et al. 1996, 1998). However, recent immunocytochemical studies using a pan-antibody that recognizes both the SERCA2a and SERCA2b isoforms revealed that the SERCA2 protein is indeed present in the somata and dendrites of dopaminergic neurons in the substantia nigra pars compacta, suggesting its involvement in somatodendritic dopamine (DA) release (Patel et al. 2009). Taken together, these expression data indicate that the SERCA2b is ubiquitously expressed in nerve cells throughout the brain, whereas SERCA2a is found almost exclusively in cerebellum (Verkhratsky 2005).
A Role for the SERCA2 in the Pathophysiology of Neuropsychiatric Disorders?
Disruption of the Ca2+ homeostasis in the brain leads to a variety of neuropsychiatric and neurodegenerative disorders. Given the prominent role of the SERCA2 pump in regulating the Ca2+ availability in the neuronal cytosol (MacLennan et al. 1985; Pozzan et al. 1994), perturbed function of this gene may result in aberrations in intracellular Ca2+-dependent molecular cascades (Berridge et al. 1998). Indeed, SERCA2-dependent Ca2+ dysregulation has been implicated in the pathophysiology of several disorders that affect cognitive function, including Darier’s disease (DD), Schizophrenia, Alzheimer’s disease (AD), and cerebral ischemia.
Darier’s Disease (DD)
DD, also known as keratosis follicularis, is an autosomal dominant skin disorder characterized by warty papules and keratotic plaques (Burge and Wilkinson 1992). DD shows almost complete penetrance and it affects between 1 in 36,000 and 1 in 100,000 individuals worldwide (Ringpfeil et al. 2001). The onset of the disease usually occurs within the second decade of life (Burge and Wilkinson 1992). The majority of DD cases present with mutations in the ATP2A2 (i.e., SERCA2) gene on chromosome 12q23-24.1. Indeed, a variety of altered-splicing, missense, nonsense, and frameshift ATP2A2 mutations have been described throughout the years. Notably, several neuropsychiatric disorders appear to be more prevalent among DD patients, including: schizophrenia, bipolar disorder (BD), epilepsy, mild mental retardation, affective psychosis, major depression disorder (MDD) (Cheour et al. 2009; Gordon-Smith et al. 2010; Jones et al. 2002; Wang et al. 2002). Interestingly, a population-based study recently reported that DD patients are four times more likely to suffer from BD and two times more likely to develop schizophrenia, as compared to the general population (Cederlöf et al. 2015). Indeed, an early study by Jacobsen et al. (1999) revealed 17 ATP2A2 mutations in affected individuals, all of which were correlated with neuropsychiatric disorders. Remarkably, mutations on the ATP-binding domain of the SERCA2 pump were correlated with BD and dysthymia (Jacobsen et al. 1999). Additionally, frameshift and missense mutations on the hinge domain were related to MDD and BD, whereas mutations on the transmembrane helices were linked to epilepsy, MDD, and mental retardation (Jacobsen et al. 1999). A later study by Ringpfeil et al. (2001) identified 14 additional heterozygous mutations in the ATPA2 gene among DD patients. Notably all of these mutations were located in highly conserved regions amid all SERCA pumps of various species (MacLennan et al. 1985), indicating their functional importance; most of the mutations identified were missense and affected the stalk, phosphorylation, hinge or transduction domains, as well as the transmembrane M6/M7 helices loop. Interestingly, it was also observed that the severity of symptoms was associated with the type of mutation; in-frame deletions in the stalk domain and a missense mutation in the transduction domain resulted to the most severe DD cases, characterized by concurrent mental disorders and vegetative growth. According to the study, depressive phenotypes were identified in patients carrying four distinct missense mutations in the transduction and phosphorylation domains, as well as the M7 helix and two in-frame deletions in the S1 helix. Moreover, schizophrenia and epilepsy were observed in individuals carrying a missense mutation in the phosphorylation domain (Ringpfeil et al. 2001). Additional case reports have confirmed that missense mutations in the stalk domain of SERCA2 are associated with schizophrenic symptoms in DD patients (Takeichi et al. 2016). In the same year, Nakamura et al. (2016) identified a heterozygous altered-splicing mutation in the acceptor site of SERCA2 associated with BD in a DD patient (Nakamura et al. 2016). Another study conducted by Noda et al. (2016) indicated that ATPA2 mutations are causally related to psychosis observed in schizophrenia and BD in DD patients; a significantly higher number of likely gene-disrupting mutations was reported in DD patients with comorbid psychosis than without (Noda et al. 2016). Most importantly, a major genome-wide association study (GWAS) array conducted in 2014, confirmed the association between schizophrenia and ATPA2, further supporting the notion that the psychosis observed in individuals affected with DD is the aftermath of the pleiotropic effect of ATPA2 mutations (Schizophrenia_Working_Group_of_the_Psychiatric_Genomics-Consortium 2014). Notably the co-occurrence of skin lesions with debilitating neuropsychiatric symptoms in DD patients possibly reflects the pleiotropic functions of SERCA2 in the skin and the brain, two ectoderm-derived organs.
Schizophrenia and the 22q11 Deletion Syndrome
Schizophrenia is a debilitating heterogeneous neuropsychiatric disorder that affects approximately 1% of the general population. The most common schizophrenia-related microdeletion, known as 22q11 deletion syndrome (22q11DS) or DiGeorge syndrome, has an incidence of 1/4000–1/6000 live births (Bassett et al. 2011; Botto et al. 2003; Chow et al. 2006; Oskarsdottir et al. 2004; Pulver et al. 1994). The majority of this multigene deletion syndrome cases are attributed to de novo microdeletions on the 22q11.21–22q11.23 chromosomal region (McDonald-McGinn et al. 2001; Scambler 2000; Schreiner et al. 2013), leading to haploinsufficiency of multiple genes (Devaraju et al. 2017; Ellegood et al. 2014; Karpinski et al. 2014; Mukai et al. 2015; Papangeli and Scambler 2013; Scambler 2000; Shi and Wang 2018; Yagi et al. 2003). Children with 22q11DS present with mild to moderate cognitive defects and learning disabilities, and the cognitive functions further deteriorate with aging (Bearden et al. 2001; Eliez et al. 2000; Gothelf et al. 2007; Rauch et al. 2006; Swillen et al. 2000). It is well established that 22q11DS accounts for 1% of all schizophrenia cases, while 25% of 22q11DS patients develop schizophrenia or a psychosis-related disorder by adulthood (Bassett and Chow 2008; Fung et al. 2010; Green et al. 2009; Jonas et al. 2014; Karayiorgou et al. 2010; Schneider et al. 2014). Moreover, growing evidence suggests that 22q11DS- and non-deleted (ND)-psychosis are comparable, presenting with similar age-onset, prevalence, symptomatology, global functioning, and comorbidity (Bassett et al. 2003; Tang et al. 2017).
Intriguingly, preclinical and clinical data suggest a role for SERCA2 in the generation of cognitive symptoms in schizophrenia. A mouse model of schizophrenia-predisposing 22q11DS, the Df(16)1/+ mouse, was reported to present marked deficits in hippocampus-dependent spatial memory, assessed in the Morris Water Maze, that were accompanied by enhanced LTP at the Schaffer collateral CA3–CA1 hippocampal synapses (Earls et al. 2010). These neurobehavioral alterations were attributed to alterations in presynaptic glutamate release that were brought about by an increase in presynaptic SERCA2 expression, altering Ca2+ kinetics in the axon terminals (Earls et al. 2010). Alterations of SERCA2 levels were not confined to the hippocampus; both the cortex and cerebellum were also found to express greater levels of SERCA2 in Df(16)1/+ mice (Earls et al. 2012). Notably, SERCA2 levels were unaltered in non-neuronal tissues (e.g., liver), indicating that the reported elevations of SERCA2 protein levels are brain-specific. Interestingly, in a follow-up study the same group reported that SERCA2 upregulation in the brain of Df(16)1/+ mice was due to loss of two microRNAs (i.e., miR-25 and miR-185) that maintain normal synaptic SERCA2 levels (Earls et al. 2012). Upon depletion of miR-25 and miR-185, SERCA2 rises to abnormal levels, resulting in aberrations of presynaptic Ca2+ turnover, and high levels of glutamate release during the sustained neuronal activity that is required for induction of LTP at excitatory synapses (Earls and Zakharenko 2014). Most importantly, increased levels of SERCA2 have also been observed in the hippocampus and the prefrontal cortex of schizophrenic patients post-mortem, strongly supporting the notion that deregulation of SERCA2 function in neural circuits implicated in the regulation of cognition may affect neuronal synaptic Ca2+ dynamics and lead to cognitive deficits observed in schizophrenia and other neuropsychiatric disorders (Earls et al. 2012).
Alzheimer’s Disease (AD)
AD is the most prevalent neurodegenerative disorder comprising approximately 60–70% of all dementia cases (Reitz et al. 2011). Clinically, AD is characterized by detrimental neuronal and synaptic loss and subsequent progressive loss of cognitive functions. Most cases of AD are not hereditary; however, a small number of early-onset cases appear to have a very strong genetic component. Most of these cases are attributed to mutations in the presenilin (PS1 and PS2) genes. Presenilin is involved in the proteolytic cleavage of the transmembrane amyloid precursor protein (APP) and the formation of toxic amyloid beta (Aβ) peptides that accumulate in the extracellular space to form the amyloid plaques, a neurobiological hallmark of AD. Under physiological conditions, PS1 and PS2 are highly conserved integral membranous proteins that mainly localize to the ER. Mutations in these genes have been detected in early-onset familial AD cases, affecting proper APP cleavage to form Aβ peptides, and overfilling the ER causing elevation of intracellular Ca2+ signals and attenuated capacitative Ca2+ entry (LaFerla 2002; Leissring et al. 2000; Lopez et al. 2008; Supnet et al. 2006; Bezprozvanny and Mattson 2008; Bojarski et al. 2008; Campion et al. 1995). Interestingly, ER overfilling is the first clinical indication of presenilin mutations, an event that could be attributed either to overactivation of SERCA or attenuation of Ca2+ leakage, leading to elevated secretion of Aβ (Cheung et al. 2008; Green et al. 2008; Green and LaFerla 2008; Tu et al. 2006).
As suggested in recent excellent reviews, the intricate interplay between Ca2+ signaling, amyloid metabolism, synaptic transmission, and plasticity, may contribute to the Ca2+ dyshomeostasis observed in AD (Corona et al. 2011; Woods and Padmanabhan 2012). This complex interaction is believed to cause major remodeling of the neuronal Ca2+ network, leading to neuronal cell death and cognitive decline (Khachaturian 1989; LaFerla 2002; Shankar et al. 2007; Thibault et al. 2007). Indeed, growing evidence suggests an intricate interaction between Ca2+, APP, and Aβ in attuning synaptic transmission and plasticity (Cirrito et al. 2003; Kamenetz et al. 2003). Furthermore, it is suggested that pathologically increased Aβ synaptic levels may impair hippocampal synaptic transmission (Abramov et al. 2009). Increased intraneuronal Ca2+ levels have also been associated with the hyperphosphorylation of TAU and neuronal death (LaFerla 2002). As PS mutations account for approximately 90% of all AD-causative mutations, their effect on SERCA-dependent ER-Ca2+ dynamics has been investigated in several studies. In vitro experiments in murine fibroblasts and human neuroblastoma cell lines suggest that the PS1 holoprotein may form a complex with the SERCA2 channel, and thus participate in the regulation of intracellular Ca2+ homeostasis (Jin et al. 2010). Indeed, studies in both mammalian cell lines and Xenopus laevis oocytes showed that presenilins physically associate with SERCA2 and are required for proper functioning of SERCA2 activity (Green et al. 2008). On the other hand, SERCA2 was also found to modulate Aβ peptide formation, as part of APP processing occurs in the ER; SERCA2b overexpression in CHO cells resulted in an increase in Aβ40 levels, whereas genetic and/or pharmacological ablation of SERCA2b induced a significant decrease in both Aβ40 and Aβ42 levels (Green et al. 2008). Additional in vitro studies in which familial AD-associated PS2 mutations were introduced in human neuroblastoma cells (SH-SY5Y), embryonic cells (HEK293), HeLa cells, and fibroblasts, resulted in reduced SERCA2b activity and a subsequent partial depletion of intracellular Ca2+ stores, confirming the critical role of presenilins on SERCA activity (Brunello et al. 2009; Zatti et al. 2004). SERCA2 activity is also believed to be regulated by the Ca2+ homeostasis modulator 1 (CALHM1), an abundant ER membrane protein. Polymorphisms in CALHM1 have been associated with sporadic AD cases, by increasing Aβ40 and Aβ42 protein levels (Dreses-Werringloer et al. 2008). Intriguingly, it was also recently shown that CALHM1 induces ER stress by decreasing the affinity of the SERCA2 pump for Ca2+, further supporting that aberrations in SERCA2 function drive Ca2+ dyshomeostasis and subsequent neuronal death in AD (Gallego-Sandin et al. 2011). A recent pull-down assay in human post-mortem brains identified SERCA2 as an APP family (FE65)-binding protein, suggesting that the interplay between FE65 and SERCA2 may affect Ca2+ homeostasis in the human brain leading to AD (Nensa et al. 2014). Nensa et al. (2014) further indicated the interaction between the two proteins with co-immunoprecipitation assays using HEK293 cells. In addition, they observed elevated SERCA2 protein levels in primary hippocampal neurons of FE65/FE65-like double knockout mice, while knock-down of FE65 in HEK293 cells resulted to increased sensitivity to a specific SERCA inhibitor, thapsigargin. The suggested mechanism of action according to this study is that upon APP cleavage by presenilins, the increased levels of free APP intracellular domains (AICD) may result to either binding onto FE65 or changing the FE65 conformation. Subsequently, the AICD/FE65 complex may interact with SERCA2, regulating SERCA2 activity and therefore Ca2+ homeostasis (Nensa et al. 2014). To the best of our knowledge, currently there are not known SERCA2 mutations associated with AD. However, SERCA2 has been shown to physically interact and/or indirectly regulate key molecular players involved in AD pathogenesis (i.e., PS1/2, APP, TAU). Given that AD may involve a chronic deregulation of Ca2+ homeostasis, gaining insights into the role of SERCA2 in the pathophysiology of AD could ultimately lead to the development of novel pharmacotherapeutic approaches aimed at restoring aberrant SERCA2 function and intracellular Ca2+ levels in an effort to combat the development and the progression of this devastating disorder.
Cerebral Ischemia and Alcoholism
Cerebral ischemia is characterized by the temporary or permanent restriction of the blood supply to brain tissue, leading to oxygen and glucose deprivation. In the aftermath, functional and structural damage is caused in different brain regions leading to what is known as stroke. Ischemic stroke is a major cause of morbidity and mortality within adults worldwide (Donnan et al. 2008). The most susceptible brain region to ischemic damage is the hippocampus, with CA1 being more vulnerable and CA3 being the least vulnerable (Kirino 2000). Notably a transient ischemic insult was reported to decrease SERCA2b mRNA levels in the hippocampal CA1 region of the gerbil brain (Xia et al. 1998). On the other hand, a recent study indicated that SERCA2b is upregulated in the CA3 neurons of the hippocampus by a hypoxia-inducible transcription factor (HIF-1α), as a neuroprotective endogenous mechanism for restoring Ca2+ homeostasis after an ischemic event (Kopach et al. 2016). Further studies using an in vitro model of cerebral ischemia (Cao et al. 2016), revealed that cerebral ischemia–reperfusion injury increased apoptotic rates and significantly enhanced the cytosolic Ca2+ concentrations at rest, with a concomitant decrease in the expression of SERCA2 (Wang et al. 2017). These data suggested that the inhibition of SERCA2 could induce the accumulation of Ca2+ in the cytosol and subsequently enhance apoptosis, supporting the tight SERCA-dependent regulation of intracellular Ca2+ flow at the early stage of apoptosis, and marking SERCA2 as a potential target for the development of future therapeutic approaches against ischemic stroke.
As cerebral ischemia and alcoholism have been linked throughout the years, the effects of chronic ethanol consumption to SERCA2b levels in the brain have also been studied. It has been established that both the hippocampus and the cerebellum are sensitive to ethanol, demonstrating neuronal cell dysfunction after chronic ethanol administration (Walker et al. 1980, 1981). Interestingly, even though the CA1 region of the hippocampus is sensitive to both ethanol and ischemia, chronic ethanol administration has no effect on the hippocampal SERCA2b mRNA levels (Coyle 1978; Goldman et al. 1973; Xia et al. 1998). However, chronic ethanol administration has been reported to cause a significant decrease in SERCA2b mRNA levels in the cerebellar Purkinje neurons and granular cell layer, as well as decreases in SERCA2b densities within the dendritic arbor of Purkinje neurons (Cassidy et al. 2013; Xia et al. 1998). Indeed, chronic ethanol administration (40 weeks) in rats was found to cause dilation of the smooth endoplasmic reticulum (SER) in the dendrites of cerebellar Purkinje neurons (PN) accompanied by decreased SERCA2b levels that possibly underlies ethanol-induced decreases in the total number of dendritic PN synapses and cerebellum-dependent balance deficits (Cassidy et al. 2013; Dlugos 2006a, b, 2008).
As SERCA2b is ubiquitously expressed in the brain, and predominately expressed in the Purkinje neurons, it is possible that Ca2+-mediated neuroprotection and ischemia may be associated with alterations in SERCA2b expression and function. To support this notion, studies have revealed that alcohol abuse may alter Ca2+ homeostasis, resulting to ER stress, caused by the overloading or depletion of Ca2+ in the ER (Dlugos 2015; Garthwaite et al. 1992; Lovinger 1993; Nagy 2000; Paschen 2003). Interestingly, as extensively conversed by Dlugos (2015), the mechanism underlying ethanol-induced decrease in the PN dendritic synapses could include the elevation of dendritic resting Ca2+ levels caused by the ethanol-induced decreased SERCA2b expression. The decline in the Ca2+ levels of the SER would then deregulate Ca2+ homeostasis, inducing ER stress and subsequent formation of degenerating bodies in the dendrites. Ultimately, SER would collapse leading to the deletion of the terminal segment at the dendritic branch point (Dlugos 2015).
Conclusions
Ca2+ is a crucial component of neuronal cell function and survival. Apart from regulating the electrophysiological properties of the neurons, it also serves as a prominent second messenger, regulating a constellation of intracellular molecular cascades. Furthermore, SERCA2 isoforms are major molecular players involved in maintaining intracellular Ca2+ balance in the brain. Even though only DD has been directly linked with SERCA2 mutations, other brain disorders present with Ca2+ dyshomeostasis due to alterations in SERCA2 expression and/or function, including Alzheimer’s disease, schizophrenia, and cerebral ischemia. Despite the fact that the SERCA2 expression pattern in the CNS was established more than two decades ago, the regulatory mechanisms that govern neuronal SERCA2b function have not been characterized. To the best of our knowledge, this is the first literature review that specifically highlights the important role of the SERCA2 in regulating Ca2+ homeostasis in the CNS. Future research should specifically address how SERCA2 expression and/or function is altered in different brain disorders, as well as which SERCA2-dependent Ca2+-regulatory pathways operate in the different neural circuits. Overall, advancing knowledge on the role that SERCA2 plays in maintaining neuronal Ca2+ homeostasis may ultimately lead to the development of safer and more effective pharmacotherapies to combat debilitating neuropsychiatric disorders.
References
Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I (2009) Amyloid-β as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci 12:1567
Artola A, Singer W (1993) Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci 16:480–487
Baba-Aissa F, Raeymaekers L, Wuytack F, De Greef C, Missiaen L, Casteels R (1996) Distribution of the organellar Ca2+ transport ATPase SERCA2 isoforms in the cat brain. Brain Res 743:141–153
Baba-Aissa F, Raeymaekers L, Wuytack F, Dode L, Casteels R (1998) Distribution and isoform diversity of the organellar Ca2+ pumps in the brain. Mol Chem Neuropathol 33:199–208. https://doi.org/10.1007/bf02815182
Baker K, Edwards T, Rickard N (2008) Inhibition of mGluR1 and IP 3 Rs impairs long-term memory formation in young chicks. Neurobiol Learn Mem 90:269–274
Bassett AS, Chow EW (2008) Schizophrenia and 22q11.2 deletion syndrome. Curr Psychiatry Rep 10:148–157
Bassett AS, Chow EW, AbdelMalik P, Gheorghiu M, Husted J, Weksberg R (2003) The schizophrenia phenotype in 22q11 deletion syndrome. Am J Psychiatry 160:1580–1586
Bassett AS et al (2011) Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr 159(332–339):e331. https://doi.org/10.1016/j.jpeds.2011.02.039
Bearden CE et al (2001) The neurocognitive phenotype of the 22q11.2 deletion syndrome: selective deficit in visual-spatial memory. J Clin Exp Neuropsychol 23:447–464
Berridge MJ, Bootman MD, Lipp P (1998) Calcium—a life and death signal. Nature 395:645–648. https://doi.org/10.1038/27094
Berridge MJ, Lipp P, Bootman MD (2000) The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1:11–21. https://doi.org/10.1038/35036035
Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4:517–529
Bezprozvanny I, Mattson MP (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 31:454–463. https://doi.org/10.1016/j.tins.2008.06.005
Bobe R et al (1998) Expression of two isoforms of the third sarco/endoplasmic reticulum Ca2+ ATPase (SERCA3) in platelets. Possible recognition of the SERCA3b isoform by the PL/IM430 monoclonal antibody. FEBS Lett 423:259–264
Bobe R et al (2004) Identification, expression, function, and localization of a novel (sixth) isoform of the human sarco/endoplasmic reticulum Ca2+ ATPase 3 gene. J Biol Chem 279:24297–24306
Bojarski L, Herms J, Kuznicki J (2008) Calcium dysregulation in Alzheimer’s disease. Neurochem Int 52:621–633. https://doi.org/10.1016/j.neuint.2007.10.002
Bononi A et al (2013) Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ 20:1631
Botto LD et al (2003) A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics 112:101–107
Brandl CJ, Green NM, Korczak B, MacLennan DH (1986) Two Ca2+ ATPase genes: homologies and mechanistic implications of deduced amino acid sequences. Cell 44:597–607
Brandl CJ, deLeon S, Martin DR, MacLennan DH (1987) Adult forms of the Ca2+ ATPase of sarcoplasmic reticulum. Expression in developing skeletal muscle. J Biol Chem 262:3768–3774
Brini M, Carafoli E (2009) Calcium pumps in health and disease. Physiol Rev 89:1341–1378. https://doi.org/10.1152/physrev.00032.2008
Brini M, Cali T, Ottolini D, Carafoli E (2014) Neuronal calcium signaling: function and dysfunction. CMLS 71:2787–2814. https://doi.org/10.1007/s00018-013-1550-7
Brini M, Carafoli E, Cali T (2017) The plasma membrane calcium pumps: focus on the role in (neuro)pathology. Biochem Biophys Res Commun 483:1116–1124. https://doi.org/10.1016/j.bbrc.2016.07.117
Brunello L, Zampese E, Florean C, Pozzan T, Pizzo P, Fasolato C (2009) Presenilin-2 dampens intracellular Ca(2+) stores by increasing Ca(2+) leakage and reducing Ca(2+) uptake. J Cell Mol Med 13:3358–3369. https://doi.org/10.1111/j.1582-4934.2009.00755.x
Burdakov D, Petersen OH, Verkhratsky A (2005) Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium 38:303–310
Burge SM, Wilkinson JD (1992) Darier–White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol 27:40–50
Burk SE, Lytton J, MacLennan DH, Shull GE (1989) cDNA cloning, functional expression, and mRNA tissue distribution of a third organellar Ca2+ pump. J Biol Chem 264:18561–18568
Camacho P, Lechleiter JD (1993) Increased frequency of calcium waves in Xenopus laevis oocytes that express a calcium-ATPase. Science 260:226–229
Campbell AM, Kessler PD, Fambrough DM (1992) The alternative carboxyl termini of avian cardiac and brain sarcoplasmic reticulum/endoplasmic reticulum Ca(2+)-ATPases are on opposite sides of the membrane. J Biol Chem 267:9321–9325
Campbell AM, Wuytack F, Fambrough DM (1993) Differential distribution of the alternative forms of the sarcoplasmic/endoplasmic reticulum Ca(2+)-ATPase, SERCA2b and SERCA2a, in the avian brain. Brain Res 605:67–76
Campion D et al (1995) Mutations of the presenilin I gene in families with early-onset Alzheimer’s disease. Hum Mol Genet 4:2373–2377
Cao Y, Zhang L, Sun S, Yi Z, Jiang X, Jia D (2016) Neuroprotective effects of syringic acid against OGD/R-induced injury in cultured hippocampal neuronal cells. Int J Mol Med 38:567–573
Carafoli E (2003) The calcium-signalling saga: tap water and protein crystals. Nat Rev Mol Cell Biol 4:326–332. https://doi.org/10.1038/nrm1073
Carafoli E, Brini M (2000) Calcium pumps: structural basis for and mechanism of calcium transmembrane transport. Curr Opin Chem Biol 4:152–161
Carafoli E, Santella L, Branca D, Brini M (2001) Generation, control, and processing of cellular calcium signals. Crit Rev Biochem Mol Biol 36:107–260. https://doi.org/10.1080/20014091074183
Cassidy LL, Dlugos FF, Dlugos CA (2013) Time course of SERCA 2b and calreticulin expression in Purkinje neurons of ethanol-fed rats with behavioral correlates. Alcohol Alcohol 48:667–678. https://doi.org/10.1093/alcalc/agt062
Cederlöf M et al (2015) The association between Darier disease, bipolar disorder, and schizophrenia revisited: a population-based family study. Bipolar Disord 17:340–344. https://doi.org/10.1111/bdi.12257
Cheour M, Zribi H, Abdelhak S, Drira S, Ben Osman A (2009) Darier’s disease: an evaluation of its neuropsychiatric component. L’Encephale 35:32–35. https://doi.org/10.1016/j.encep.2007.09.009
Cheung K-H et al (2008) Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58:871–883
Chow EW, Watson M, Young DA, Bassett AS (2006) Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophr Res 87:270–278
Cirrito JR et al (2003) In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-β metabolism and half-life. J Neurosci 23:8844–8853
Clapham DE (1995) Calcium signaling. Cell 80:259–268
Corona C, Pensalfini A, Frazzini V, Sensi S (2011) New therapeutic targets in Alzheimer’s disease: brain deregulation of calcium and zinc. Cell Death Dis 2:e176
Coyle P (1978) Spatial features of the rat hippocampal vascular system. Exp Neurol 58:549–561
Csordás G et al (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174:915
Csordás G et al (2010) Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell 39:121–132
Dahl R (2017) A new target for Parkinson’s disease: small molecule SERCA activator CDN1163 ameliorates dyskinesia in 6-OHDA-lesioned rats. Bioorg Med Chem 25:53–57. https://doi.org/10.1016/j.bmc.2016.10.008
Dally S et al (2006) Ca2+-ATPases in non-failing and failing heart: evidence for a novel cardiac sarco/endoplasmic reticulum Ca2+-ATPase 2 isoform (SERCA2c). Biochem J 395:249–258. https://doi.org/10.1042/bj20051427
Dally S, Corvazier E, Bredoux R, Bobe R, Enouf J (2009) Multiple and diverse coexpression, location, and regulation of additional SERCA2 and SERCA3 isoforms in nonfailing and failing human heart. J Mol Cell Cardiol 48:633–644. https://doi.org/10.1016/j.yjmcc.2009.11.012
Dally S, Corvazier E, Bredoux R, Bobe R, Enouf J (2010) Multiple and diverse coexpression, location, and regulation of additional SERCA2 and SERCA3 isoforms in nonfailing and failing human heart. J Mol Cell Cardiol 48:633–644. https://doi.org/10.1016/j.yjmcc.2009.11.012
Devaraju P et al (2017) Haploinsufficiency of the 22q11.2 microdeletion gene Mrpl40 disrupts short-term synaptic plasticity and working memory through dysregulation of mitochondrial calcium. Mol Psychiatry 22:1313
Dlugos CA (2006a) Ethanol-related smooth endoplasmic reticulum dilation in Purkinje dendrites of aging rats. Alcohol Clin Exp Res 30:883–891
Dlugos CA (2006b) Smooth endoplasmic reticulum dilation and degeneration in Purkinje neuron dendrites of aging ethanol-fed female rats. Cerebellum 5:155–162
Dlugos CA (2008) Ethanol-related increases in degenerating bodies in the Purkinje neuron dendrites of aging rats. Brain Res 1221:98–107
Dlugos CA (2015) Ethanol-induced alterations in Purkinje neuron dendrites in adult and aging rats: a review. Cerebellum 14:466–473
Dode L, De Greef C, Mountian I, Attard M, Town MM, Casteels R, Wuytack F (1998) Structure of the human sarco/endoplasmic reticulum Ca2+-ATPase 3 gene promoter analysis and alternative splicing of the SERCA3 pre-mRNA. J Biol Chem 273:13982–13994
Dode L, Andersen JP, Leslie N, Dhitavat J, Vilsen B, Hovnanian A (2003) Dissection of the functional differences between sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) 1 and 2 isoforms and characterization of Darier disease (SERCA2) mutants by steady-state and transient kinetic analyses. J Biol Chem 278:47877–47889
Donnan GA, Fisher M, Macleod M, Davis SM (2008) Stroke. Lancet 371:1612–1623. https://doi.org/10.1016/s0140-6736(08)60694-7
Dreses-Werringloer U et al (2008) A polymorphism in CALHM1 influences Ca2+ homeostasis, abeta levels, and Alzheimer’s disease risk. Cell 133:1149–1161. https://doi.org/10.1016/j.cell.2008.05.048
Duchen MR (1999) Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. J Physiol 516:1–17
Earls LR, Zakharenko SS (2014) A Synaptic function approach to investigating complex psychiatric diseases. Neuroscientist 20:257–271. https://doi.org/10.1177/1073858413498307
Earls LR, Bayazitov IT, Fricke RG, Berry RB, Illingworth E, Mittleman G, Zakharenko SS (2010) Dysregulation of presynaptic calcium and synaptic plasticity in a mouse model of 22q11 deletion syndrome. J Neurosci 30:15843–15855. https://doi.org/10.1523/jneurosci.1425-10.2010
Earls LR, Fricke RG, Yu J, Berry RB, Baldwin LT, Zakharenko SS (2012) Age-dependent microRNA control of synaptic plasticity in 22q11 deletion syndrome and schizophrenia. J Neurosci 32:14132–14144. https://doi.org/10.1523/jneurosci.1312-12.2012
Eliez S et al (2000) Young children with Velo-Cardio-Facial syndrome (CATCH-22).Psychological and language phenotypes. Eur Child Adolesc Psychiatry 9:109–114
Ellegood J et al (2014) Neuroanatomical phenotypes in a mouse model of the 22q11.2 microdeletion. Mol Psychiatry 19:99–107. https://doi.org/10.1038/mp.2013.112
Fucile S (2004) Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium 35:1–8
Fung WL, McEvilly R, Fong J, Silversides C, Chow E, Bassett A (2010) Elevated prevalence of generalized anxiety disorder in adults with 22q11.2 deletion syndrome. Am J Psychiatry 167:998. https://doi.org/10.1176/appi.ajp.2010.09101463
Gallego-Sandin S, Alonso MT, Garcia-Sancho J (2011) Calcium homoeostasis modulator 1 (CALHM1) reduces the calcium content of the endoplasmic reticulum (ER) and triggers ER stress. Biochem J 437:469–475. https://doi.org/10.1042/bj20110479
Garthwaite G, Hajos F, Garthwaite J (1992) Morphological response of endoplasmic reticulum in cerebellar Purkinje cells to calcium deprivation. Neuroscience 48:681–688
Gelebart P, Martin V, Enouf J, Papp B (2003) Identification of a new SERCA2 splice variant regulated during monocytic differentiation. Biochem Biophys Res Commun 303:676–684
Goldman H, Sapirstein LA, Murphy S, Moore J (1973) Alcohol and regional blood flow in brains of rats. Proc Soc Exp Biol Med 144:983–988
Gordon-Smith K, Jones LA, Burge SM, Munro CS, Tavadia S, Craddock N (2010) The neuropsychiatric phenotype in Darier disease. Br J Dermatol 163:515–522. https://doi.org/10.1111/j.1365-2133.2010.09834.x
Gothelf D, Penniman L, Gu E, Eliez S, Reiss AL (2007) Developmental trajectories of brain structure in adolescents with 22q11.2 deletion syndrome: a longitudinal study. Schizophr Res 96:72–81
Green KN, LaFerla FM (2008) Linking calcium to Aβ and Alzheimer’s disease. Neuron 59:190–194
Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM (2008) SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Gen Physiol 132:i1. https://doi.org/10.1085/jgp1322oia1
Green T et al (2009) Psychiatric disorders and intellectual functioning throughout development in velocardiofacial (22q11.2 deletion) syndrome. J Am Acad Child Adolesc Psychiatry 48:1060–1068
Grienberger C, Konnerth A (2012) Imaging calcium in neurons neuron 73:862–885. https://doi.org/10.1016/j.neuron.2012.02.011
Guerini D (1998) The Ca2+ pumps and the Na+/Ca2+ exchangers. Biometals 11:319–330
Gunteski-Hamblin AM, Greeb J, Shull GE (1988) A novel Ca2+ pump expressed in brain, kidney, and stomach is encoded by an alternative transcript of the slow-twitch muscle sarcoplasmic reticulum Ca-ATPase gene. Identification of cDNAs encoding Ca2+ and other cation-transporting ATPases using an oligonucleotide probe derived from the ATP-binding site. J Biol Chem 263:15032–15040
Hao L, Rigaud J-L, Inesi G (1994) Ca2+/H+ countertransport and electrogenicity in proteoliposomes containing erythrocyte plasma membrane Ca-ATPase and exogenous lipids. J Biol Chem 269:14268–14275
Hasselbach W, Makinose M (1961) The calcium pump of the “relaxing granules” of muscle and its dependence on ATP-splitting. Biochem Z 333:518–528
Hayashi T, Rizzuto R, Hajnoczky G, Su T-P (2009) MAM: more than just a housekeeper. Trends Cell Biol 19:81–88. https://doi.org/10.1016/j.tcb.2008.12.002
Higgins ER, Cannell MB, Sneyd J (2006) A buffering SERCA pump in models of calcium dynamics. Biophys J 91:151–163. https://doi.org/10.1529/biophysj.105.075747
Jacobsen NJ et al (1999) ATP2A2 mutations in Darier’s disease and their relationship to neuropsychiatric phenotypes. Hum Mol Genet 8:1631–1636
Jin H, Sanjo N, Uchihara T, Watabe K, St George-Hyslop P, Fraser PE, Mizusawa H (2010) Presenilin-1 holoprotein is an interacting partner of sarco endoplasmic reticulum calcium-ATPase and confers resistance to endoplasmic reticulum stress. JAD 20:261–273. https://doi.org/10.3233/jad-2010-1360
Jonas RK, Montojo CA, Bearden CE (2014) The 22q11.2 deletion syndrome as a window into complex neuropsychiatric disorders over the lifespan. Biol Psychiatry 75:351–360. https://doi.org/10.1016/j.biopsych.2013.07.019
Jones I, Jacobsen N, Green EK, Elvidge GP, Owen MJ, Craddock N (2002) Evidence for familial cosegregation of major affective disorder and genetic markers flanking the gene for Darier’s disease. Mol Psychiatry 7:424–427. https://doi.org/10.1038/sj.mp.4000989
Kamenetz F et al (2003) APP processing and synaptic function. Neuron 37:925–937
Karayiorgou M, Simon TJ, Gogos JA (2010) 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nat Rev Neurosci 11:402 https://doi.org/10.1038/nrn2841. https://www.nature.com/articles/nrn2841#supplementary-information
Karpinski BA, Maynard TM, Fralish MS, Nuwayhid S, Zohn IE, Moody SA, LaMantia AS (2014) Dysphagia and disrupted cranial nerve development in a mouse model of DiGeorge (22q11) deletion syndrome. Dis Models Mech 7:245–257. https://doi.org/10.1242/dmm.012484
Kawamoto EM, Vivar C, Camandola S (2012) Physiology and pathology of calcium signaling in the brain. Front Pharmacol 3:61
Khachaturian ZS (1989) Calcium, membranes, aging and Alzheimer’s disease: introduction and overview. Ann N Y Acad Sci 568:1–4. https://doi.org/10.1111/j.1749-6632.1989.tb12485.x
Kimura T et al (2005) Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum Mol Genet 14:2189–2200. https://doi.org/10.1093/hmg/ddi223
Kirino T (2000) Delayed neuronal death. Neuropathology 20(Suppl):S95–S97
Kopach O, Maistrenko A, Lushnikova I, Belan P, Skibo G, Voitenko N (2016) HIF-1alpha-mediated upregulation of SERCA2b: the endogenous mechanism for alleviating the ischemia-induced intracellular Ca(2+) store dysfunction in CA1 and CA3 hippocampal neurons. Cell Calcium 59:251–261. https://doi.org/10.1016/j.ceca.2016.02.014
Korczak B, Zarain-Herzberg A, Brandl CJ, Ingles CJ, Green NM, MacLennan DH (1988) Structure of the rabbit fast-twitch skeletal muscle Ca2+-ATPase gene. J Biol Chem 263:4813–4819
Kranias EG, Hajjar RJ (2012) Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res 110:1646–1660. https://doi.org/10.1161/CIRCRESAHA.111.259754
LaFerla FM (2002) Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci 3:862–872. https://doi.org/10.1038/nrn960
Lee C-H, Poburko D, Kuo K-H, Seow CY, van Breemen C (2002) Ca2+ oscillations, gradients, and homeostasis in vascular smooth muscle. Am J Heart Circ Physiol 282:H1571–H1583
Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM (2000) Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol 149:793–798
Lopez JR, Lyckman A, Oddo S, LaFerla FM, Querfurth HW, Shtifman A (2008) Increased intraneuronal resting [Ca2+] in adult Alzheimer’s disease mice. J Neurochem 105:262–271
Lovinger DM (1993) Excitotoxicity and alcohol-related brain damage. Alcohol Clin Exp Res 17:19–27
Lyons MR, West AE (2011) Mechanisms of specificity in neuronal activity-regulated gene transcription. Prog Neurobiol 94:259–295
Lytton J, MacLennan DH (1988) Molecular cloning of cDNAs from human kidney coding for two alternatively spliced products of the cardiac Ca2+-ATPase gene. J Biol Chem 263:15024–15031
Lytton J, Zarain-Herzberg A, Periasamy M, MacLennan DH (1989) Molecular cloning of the mammalian smooth muscle sarco(endo)plasmic reticulum Ca2+-ATPase. J Biol Chem 264:7059–7065
Lytton J, Westlin M, Burk SE, Shull GE, MacLennan DH (1992) Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J Biol Chem 267:14483–14489
MacLennan DH (1970) Purification and properties of an adenosine triphosphatase from sarcoplasmic reticulum. J Biol Chem 245:4508–4518
MacLennan DH, Brandl CJ, Korczak B, Green NM (1985) Amino-acid sequence of a Ca2+ + Mg2+-dependent ATPase from rabbit muscle sarcoplasmic reticulum, deduced from its complementary DNA sequence. Nature 316:696–700
Martin V, Bredoux R, Corvazier E, van Gorp R, Kovàcs T, Gélébart P, Enouf J (2002) Three novel sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) 3 isoforms expression, regulation, and function of the members of the SERCA3 family. J Biol Chem 277:24442–24452
McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, Finucane B, Driscoll DA, Emanuel BS, Zackai EH (2001) Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet Med 3:23–29. https://doi.org/10.1097/00125817-200101000-00006
Miller KK, Verma A, Snyder SH, Ross CA (1991) Localization of an endoplasmic reticulum calcium ATPase mRNA in rat brain by in situ hybridization. Neuroscience 43:1–9
Møller JV, Olesen C, Jensen A-ML, Nissen P (2005) The structural basis for coupling of Ca2+ transport to ATP hydrolysis by the sarcoplasmic reticulum Ca2+ -ATPase. J Bioenerg Biomembr 37:359–364
Morita M, Kudo Y (2010) Growth factors upregulate astrocyte [Ca2+]i oscillation by increasing SERCA2b expression. Glia 58:1988–1995. https://doi.org/10.1002/glia.21067
Mukai J et al (2015) Molecular substrates of altered axonal growth and brain connectivity in a mouse model of schizophrenia. Neuron 86:680–695. https://doi.org/10.1016/j.neuron.2015.04.003
Mulkey RM, Malenka RC (1992) Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 9:967–975
Nagy J (2000) Alcohol dependence at the cellular level: effects of ethanol on calcium homeostasis of IM-9 human lymphoblast cells. J Stud Alcohol 61:225–231
Nakamura T, Kazuno AA, Nakajima K, Kusumi I, Tsuboi T, Kato T (2016) Loss of function mutations in ATP2A2 and psychoses: a case report and literature survey. Psychiatry Clin Neurosci 70:342–350. https://doi.org/10.1111/pcn.12395
Neher E, Sakaba T (2008) Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59:861–872
Nensa FM et al (2014) Amyloid beta a4 precursor protein-binding family B member 1 (FE65) interactomics revealed synaptic vesicle glycoprotein 2A (SV2A) and sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2) as new binding proteins in the human brain. Mol Cell Proteom 13:475–488. https://doi.org/10.1074/mcp.m113.029280
Noda K et al (2016) Novel and recurrent ATP2A2 mutations in Japanese patients with Darier’s disease. Nagoya J Med Sci 78:485–492. https://doi.org/10.18999/nagjms.78.4.485
Olesen C, Sørensen TL-M, Nielsen RC, Møller JV, Nissen P (2004) Dephosphorylation of the calcium pump coupled to counterion occlusion. Science 306:2251–2255
Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium–apoptosis link. Nat Rev Mol Cell Biol 4:552–565
Oskarsdottir S, Vujic M, Fasth A (2004) Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch Dis Child 89:148–151
Papangeli I, Scambler P (2013) The 22q11 deletion: DiGeorge and velocardiofacial syndromes and the role of TBX1. Wiley Interdiscip Rev 2:393–403. https://doi.org/10.1002/wdev.75
Paschen W (2003) Endoplasmic reticulum: a primary target in various acute disorders and degenerative diseases of the brain. Cell Calcium 34:365–383
Patel JC, Witkovsky P, Avshalumov MV, Rice ME (2009) Mobilization of calcium from intracellular stores facilitates somatodendritic dopamine release. J Neurosci 29:6568–6579. https://doi.org/10.1523/jneurosci.0181-09.2009
Periasamy M, Kalyanasundaram A (2007) SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve 35:430–442
Plessers L, Eggermont JA, Wuytack F, Casteels R (1991) A study of the organellar Ca2(+)-transport ATPase isozymes in pig cerebellar Purkinje neurons. J Neurosci 11:650–656
Poch E, Leach S, Snape S, Cacic T, MacLennan DH, Lytton J (1998) Functional characterization of alternatively spliced human SERCA3 transcripts. Am J Physiol 275:C1449–C1458
Pozzan T, Rizzuto R, Volpe P, Meldolesi J (1994) Molecular and cellular physiology of intracellular calcium stores. Physiol Rev 74:595–636
Pulver AE et al (1994) Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. J Nerv Ment Dis 182:476–477
Rauch A et al (2006) Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet A 140:2063–2074. https://doi.org/10.1002/ajmg.a.31416
Reitz C, Brayne C, Mayeux R (2011) Epidemiology of Alzheimer disease. Nat Rev Neurol 7:137–152. https://doi.org/10.1038/nrneurol.2011.2
Ringpfeil F et al (2001) Darier disease—novel mutations in ATP2A2 and genotype-phenotype correlation. Exp Dermatol 10:19–27
Salinska EJ, Bourne RC, Rose SP (2001) Long-term memory formation in the chick requires mobilization of ryanodine-sensitive intracellular calcium stores. Neurobiol Learn Mem 75:293–302
Salvador JM, Inesi G, Rigaud J-L, Mata AM (1998) Ca2+ transport by reconstituted synaptosomal ATPase is associated with H+ countertransport and net charge displacement. J Biol Chem 273:18230–18234
Salvador JM, Berengena M, Sepulveda MR, Mata AM (2001) Distribution of the intracellular Ca(2+)-ATPase isoform 2b in pig brain subcellular fractions and cross-reaction with a monoclonal antibody raised against the enzyme isoform. J Biochem 129:621–626
Scambler PJ (2000) The 22q11 deletion syndromes. Hum Mol Genet 9:2421–2426
Schizophrenia_Working_Group_of_the_Psychiatric_Genomics-Consortium (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature 511:421–427. https://doi.org/10.1038/nature13595
Schneider M et al (2014) Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am J Psychiatry 171:627–639
Schreiner MJ, Lazaro MT, Jalbrzikowski M, Bearden CE (2013) Converging levels of analysis on a genomic hotspot for psychosis: insights from 22q11.2 deletion syndrome. Neuropharmacology 68:157–173
Schwaller B (2010) Cytosolic Ca2+ buffers. Cold Spring Harbor Perspect Biol 2:a004051
Sepulveda MR, Hidalgo-Sanchez M, Mata AM (2004) Localization of endoplasmic reticulum and plasma membrane Ca2+-ATPases in subcellular fractions and sections of pig cerebellum. Eur J Neurosci 19:542–551
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL (2007) Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27:2866–2875
Shi H, Wang Z (2018) Atypical microdeletion in 22q11 deletion syndrome reveals new candidate causative genes: a case report and literature review. Medicine 97:e9936. https://doi.org/10.1097/md.0000000000009936
Simonyi A, Schachtman TR, Christoffersen G (2005) The role of metabotropic glutamate receptor 5 in learning and memory processes. Drug News Perspect 18:353–361
Supnet C, Grant J, Westaway D, Mayne M (2006) Amyloid-β-(1-42) increases ryanodine receptor-3 expression and function in neurons of TgCRND8 mice. J Biol Chem 281:38440–38447
Swillen A, Vogels A, Devriendt K, Fryns J-P (2000) Chromosome 22q11 deletion syndrome: update and review of the clinical features, cognitive-behavioral spectrum, and psychiatric complications. Am J Med Genet A 97:128–135
Takeichi T, Sugiura K, Nakamura Y, Fujio Y, Konohana I, Akiyama M (2016) Darier’s disease complicated by schizophrenia caused by a novel ATP2A2 mutation. Acta dermatol 96:993–994. https://doi.org/10.2340/00015555-2422
Tang SX et al (2017) The psychosis SPECTRUM in 22q11.2 deletion syndrome Is comparable to that of nondeleted youth. Biol Psychiatry 82:17–25. https://doi.org/10.1016/J.BIOPSYCH.2016.08.034
Taylor CW, Tovey SC (2010) IP(3) receptors: toward understanding their activation. Cold Spring Harbor Perspect Biol 2:a004010. https://doi.org/10.1101/cshperspect.a004010
Thibault O, Gant JC, Landfield PW (2007) Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: minding the store. Aging Cell 6:307–317
Toyofuku T, Kurzydlowski K, Lytton J, MacLennan D (1992) The nucleotide binding/hinge domain plays a crucial role in determining isoform-specific Ca2+ dependence of organellar Ca (2+)-ATPases. J Biol Chem 267:14490–14496
Toyoshima C, Inesi G (2004) Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annu Rev Biochem 73:269–292. https://doi.org/10.1146/annurev.biochem.73.011303.073700
Tu H et al (2006) Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126:981–993
van Vliet AR, Verfaillie T, Agostinis P (2014) New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta 1843:2253–2262. https://doi.org/10.1016/j.bbamcr.2014.03.009
Vandecaetsbeek I et al (2009) Structural basis for the high Ca2+ affinity of the ubiquitous SERCA2b Ca2+ pump. Proc Natl Acad Sci 106:18533–18538
Verboomen H, Wuytack F, De Smedt H, Himpens B, Casteels R (1992) Functional difference between SERCA2a and SERCA2b Ca2+ pumps and their modulation by phospholamban. Biochem J 286:591–595
Verboomen H, Wuytack F, Van den Bosch L, Mertens L, Casteels R (1994) The functional importance of the extreme C-terminal tail in the gene 2 organellar Ca(2+)-transport ATPase (SERCA2a/b). Biochem J 303(3):979–984
Verkhratsky A (2005) Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev 85:201–279. https://doi.org/10.1152/physrev.00004.2004
Walker DW, Barnes DE, Zornetzer SF, Hunter BE, Kubanis P (1980) Neuronal loss in hippocampus induced by prolonged ethanol consumption in rats. Science 209:711–713
Walker DW, Hunter BE, Abraham WC (1981) Neuroanatomical and functional deficits subsequent to chronic ethanol administration in animals. Alcohol Clin Exp Res 5:267–282
Wang SL, Yang SF, Chen CC, Tsai PT, Chai CY (2002) Darier’s disease associated with bipolar affective disorder: a case report. Kaohsiung J Med Sci 18:622–626
Wang K, Chen M, Gong H, Lou Y, Gong X, Zhong X, Huang Z (2017) Calcium homeostasis disruption and endoplasmic reticulum stress mediats ischemia/reperfusion-induced PC12 cells apoptosis. Int J Clin Exp Med 10:14121–14129
Wu KD, Lee WS, Wey J, Bungard D, Lytton J (1995) Localization and quantification of endoplasmic reticulum Ca(2+)-ATPase isoform transcripts. Am J Physiol 269:C775–C784
Wuytack F, Raeymaekers L, Missiaen L (2002) Molecular physiology of the SERCA and SPCA pumps. Cell Calcium 32:279–305
Woods NK, Padmanabhan J (2012) Neuronal calcium signaling and Alzheimer’s disease. In: Islam MS (ed) Calcium signaling. Springer, Dordrecht, pp 1193–1217. https://doi.org/10.1007/978-94-007-2888-2_54
Xia J, Simonyi A, Sun GY (1998) Changes in IP3R1 and SERCA2b mRNA levels in the gerbil brain after chronic ethanol administration and transient cerebral ischemia-reperfusion Brain research. Mol Brain Res 56:22–28
Yagi H et al (2003) Role of TBX1 in human del22q11.2 syndrome. Lancet 362:1366–1373. https://doi.org/10.1016/S0140-6736(03)14632-6
Yu X, Carroll S, Rigaud J, Inesi G (1993) H+ countertransport and electrogenicity of the sarcoplasmic reticulum Ca2+ pump in reconstituted proteoliposomes. Biophys J 64:1232–1242
Zarain-Herzberg A, MacLennan D, Periasamy M (1990) Characterization of rabbit cardiac sarco (endo) plasmic reticulum Ca2(+)-ATPase gene. J Biol Chem 265:4670–4677
Zatti G, Ghidoni R, Barbiero L, Binetti G, Pozzan T, Fasolato C, Pizzo P (2004) The presenilin 2 M239I mutation associated with familial Alzheimer’s disease reduces Ca2+ release from intracellular stores. Neurobiol Dis 15:269–278. https://doi.org/10.1016/j.nbd.2003.11.002
Zhang P, Toyoshima C, Yonekura K, Green NM, Stokes DL (1998) Structure of the calcium pump from sarcoplasmic reticulum at 8-A resolution. Nature 392:835–839. https://doi.org/10.1038/33959
Zucker RS (1999) Calcium-and activity-dependent synaptic plasticity. Curr Opin Neurobiol 9:305–313
Acknowledgements
AB and CT were supported by the University of Dayton (UD) Graduate School and by the UD Office for Graduate Affairs through the Graduate Student Summer Fellowship (GSSF) Program. JS was supported by the Berry Summer Thesis Institute, the UD Honors Program, and by UD’s STEM Catalyst Grant program. EF was supported by a Biology Department Lancaster-McDougall Award, a Stander Undergraduate Research Fellowship, and a CAS Dean’s Summer Research fellowship. PMP was supported by an inaugural STEM Catalyst grant from the University of Dayton, as well as by Research Council Seed Grants (RCSG) from the University of Dayton Research Institute (UDRI). This review paper was compiled in the context of our “Neuropharmacology” (BIO496/596; Fall 2017) course at the University of Dayton. Funding sponsors had no further role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
AB and JS conducted the primary literature search and wrote the manuscript. EF and CT contributed to the final version of the manuscript. PMP formulated the concept, supervised the literature search and reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Britzolaki, A., Saurine, J., Flaherty, E. et al. The SERCA2: A Gatekeeper of Neuronal Calcium Homeostasis in the Brain. Cell Mol Neurobiol 38, 981–994 (2018). https://doi.org/10.1007/s10571-018-0583-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-018-0583-8