
Abstract
In our previous research, the formation and development of age-related cataract (ARC) is associated with DNA hypermethylation of some genes in lens epithelial cells (LECs). This study aimed to investigate the expression profile of DNA methylation- and transcriptional repression-associated genes in LECs of ARC. The expression levels of the genes were first evaluated by microarray analysis. The results were further confirmed by Quantitative Real-Time PCR (qRT-PCR) and Western blot assay. The mRNA and protein levels of 5 genes increased in LECs of ARCs compared with the controls. These data provided a global perspective on expression of DNA methylation- and transcriptional repression-associated genes. The study supports the notion that the epigenetic modification of macromolecules in LECs might contribute to ARC pathogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
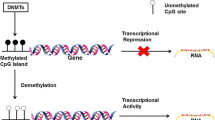
DNA methylation is the most studied epigenetic modification in many age-related diseases including age-related cataract (ARC) (Li et al. 2015; Levine et al. 2015; Palsamy et al. 2014; Li et al. 2014; Wang et al. 2015). In mammalian genome, methylation of CpG-rich regions (CpG islands) modulates gene expression without changes in the DNA sequence (Taby and Issa 2010). Gene silencing through DNA methylation occurs through the activity of DNA methyltransferases. The enzymes transfer a methyl group from S-adenosyl-l-methionine to the carbon 5 position of cytosine. There are three DNA methyltransferases DNMTs (DNMT1, DNMT3A, and DNMT3B) in human (Jin and Robertson 2013). Two main types of methyltransferase activity have been found in mammals: a de novo activity and a maintenance activity. DNMT3A and DNMT3B have been identified as de novo methyltransferases. They can methylate cytosine at CpG dinucleotides on both strands. DNMT1, the principal DNA methyltransferase in mammalian cells, acts to restore methylated cytosines at CpGs on the newly duplicated strands (Lei et al. 1996; Okano et al. 1998). However, evidences show that DNMT1 may also work together with DNMT3A and DNMT3B in de novo methyltransferase activity in certain genome in both embryonic cells and differentiated somatic cells (Ko et al. 2005; Ratnam et al. 2002). Several molecular mechanisms are thought to be responsible for this methylcytosine-mediated gene repression. Among them, the repression mediated by mCpG-binding proteins has been most extensively studied. Five mCpG-binding proteins, MBD1–4 and MeCP2 (Nan et al. 1993), have been identified in mammals and are collectively called MBD family proteins because these proteins share the mCpG-binding domain (Hendrich and Bird 1998). In the MBD family, MBD3 shows a binding activity to hemimethylated DNA. It operates as the binding proteins to mCpG sequences and transcriptional repressors (Tatematsu et al. 2000). Many interacting proteins have been reported to bind to their N-terminal region by biochemical interaction assay (Hermann et al. 2004).
Evidences suggest that the DNA methylation and histone modification are strictly linked and can reciprocally associate or interfere (Klose and Bird 2006; Vaissiere et al. 2008). Histone deacetylation is catalyzed by histone deacetylase (HDACs) including Class I HDAC, Class II HDAC, Class III HDAC, and Class IV HDAC. For example, DNMT1 directly interacts with histone modifying enzymes such as histone H3K9 methyltransferase SUV39H1, histone H3K27 methyltransferase EZH2, and histone deacetylase HDAC1 and HDAC2 (Hermann et al. 2004; Hernandez-Munoz et al. 2005). DNMT1 also interacts with methyl-CpG binding proteins such as MBD2, MBD3, and MeCP2. MeCP2 transiently interacts with the Sin3A and HDAC2 complexes. A research showed a mechanistic linkage between HDAC1/2 in HDAC-containing complexes of which SIN3A is one and DNA methylation mediated by DNMT3A during oocyte growth (Ma et al. 2015).
ARC is a complex disease with multiple genetic and environmental risk components. Epigenetic regulation is the main mechanism depending on the environmental stimulus (Jaenisch and Bird 2003). Recently, we reported that several gene expressions are regulated by DNA methylation in lens epithelium cells (LECs)(Li et al. 2014; Wang et al. 2015). However, the exact mechanism is not completely understood in the regulation. It is necessary to investigate the upstream regulation mechanisms for gene silencing by DNA hypermethylation in LECs of ARC.
In this study, we were interested in assessing whether the expression of DNA methylation- and transcriptional repression-associated genes were different in LECs between controls and ARCs. We focused on LECs, because of its role in the center of metabolic activities in lenses. Therefore, to find the key genes involved DNA methylation- and transcriptional repression-associated genes, a global approach is required. We determined the expression of the genes in LECs by a microarray.
Materials and Methods
Study Participants
This research followed the tenets of the Declaration of Helsinki and was approved by the Ethics Committee of Affiliated Hospital of Nantong University. All participants gave informed consent and all patients had a complete preoperative ophthalmologic examination. The severity of the cataracts was graded and recorded through Lens Opacities Classification System III (LOCS III)(Chylack et al. 1993). Thirty centered anterior capsules (15 males, 15 females) were collected from ARC patients without other ocular diseases. Thirty transparent control lens samples (15 males, 15 females) of the donors were obtained from the Eye Bank of Affiliated Hospital of Nantong University. There were no statistically significant differences between the two groups regarding the age and sex (Table 1).
Tissue Preparation, RNA Extraction, and Reverse Transcription
The centered anterior capsules of lens were carefully obtained by anterior continuous curvilinear capsulorhexis during cataract surgery. In the centered anterior capsules of lens, total RNA was isolated from LECs by Trizol® Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. RNA concentration was determined through a photometry at 260/280 nm. The RNA quality was assessed by the ratio of the 18/28S ribosomal band intensities in an ethidium bromide-containing 1 % agarose gel after electrophoresis. Equal amounts of RNA were reversely transcribed to cDNAs using PrimeScript® RT reagent Kit (TaKaRa, Dalian, China).
cDNA Microarray of DNA Methylation- and Transcriptional Repression-Associated Genes
The relative expression of the genes involved in DNA methylation and transcriptional repression in each of the six cDNA samples (3 controls and 3 ARCs) through TaqMan® Array Human DNA Methylation and Transcriptional Repression 96-well Plate (#4418772, Applied Biosystems, Foster City, CA). Table 2 shows all genes included in the array. Samples with equal amounts of RNA were reversely transcribed to cDNA. Then 2 μl cDNAs were diluted in ddH2O (7 μl) and Gene Expression Master Mix (10 μl) (Applied Biosystems) according to the supplier’s directions and pipetted into microarray plates. Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) was performed in ABI 7500 system (Applied Biosystems). PCR parameters were set as 95 °C for 30 s, followed by 40 cycles of 95 °C
for 5 s, 60 °C for 34 s, and 72 °C for 15 s. The microarray data were recorded as threshold cycle (ΔCT) and analyzed through DataAssist v3.01 Software (Applied Biosystems). The average expression of eight housekeeping genes (18S, GAPDH, GUSB, HPR1, B2 M, RPLP0, HMBS, and ACTB) was used for normalization of the data. After normalization, the relative expression of each gene was averaged for the three samples in each group. Fold changes in average gene expression were expressed as the difference in expression of LECs from ARCs compared with those of controls. The genes that were significantly altered with minimum 1.5-fold changes were selected for further analysis.
Quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR)
TaqMan gene expression assay probes (Applied Biosystems) were used for DNMT3B, HDAC1, HDAC4, HDAC9, and MBD3 mRNA quantification (assay ID: Hs00171876_m1, Hs02621185_ m1, Hs00195814_m1, Hs00206843_m1, and Hs00172710_m1). GAPDH (Hs99999905_m1) was used as an internal control. qRT-PCR was performed using the ABI 7500 Real-Time PCR System (Applied Biosystems). The fold change of gene expression was determined using the comparative CT method (2—ΔΔCT) and each sample was analyzed in triplicate.
Western Blot Assay
Lysates of LECs were prepared for Western blot analysis as described previously (Wang et al. 2015). After determination of its protein concentration with the Bradford assay (Bio-Rad, USA), samples with equal amounts of protein were subjected to SDS–polyacrylamide gel electrophoresis (PAGE) (100 V for 90 min) and transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA) by a transfer apparatus (Bio-Rad) at 40 mA for 8 h. Nonspecific protein binding to the membrane was blocked with blocking buffer (5 % nonfat milk, 200 mM NaCl, 50 mM Tris, 0.05 % Tween 20). The blocked membrane was then incubated with primary antibodies against DNMT3B (rabbit, 1:1000, Abcam, Inc., Cambridge, MA, USA), HDAC1 (mouse, 1:1000, Millipore, Billerica, MA), HDAC4 (goat, 1:1000, Abcam), HDAC9 (rabbit, 1:2000; Abcam), MBD3 (rabbit, 1:2000; Abcam), and GAPDH (rabbit, 1:2000; Abcam) at 4 °C for 12 h. After the membrane was washed three times with TBST (20 mM Tris, 500 mM NaCl, 0.1 % Tween 20) for 5 min each time at 28 °C, the membrane was incubated with alkaline phosphatase-conjugated secondary antibodies (1:4000; Santa Cruz, USA) for 2 h at 28 °C. Then the membrane was washed four times with TBST for 15 min each time at 28 °C. Detection was performed using an ECL chemiluminescence kit (Pierce, Rockford, IL). The film was scanned using ImageQuant software (Molecular Dynamics, Sunnyvale, CA). The gray value of each protein band was measured, and data are presented as a ratio of this value to that of GAPDH.
Statistical Analysis
Student’s t test was used to determine the difference in averages between the two groups. p Value <0.05 was considered statistically significant. Statistical analyses were performed with SPSS software (SPSS 17.0; SPSS Inc., Chicago, IL).
Results
mRNA of DNA Methylation- and Transcriptional Repression-Associated Genes in Controls Versus ARCs by Microarray
The microarray data from three pairs of ARC and control samples are presented in Fig. 1. All of the 32 probes included in the array (Table 2) were detected in each anterior lens capsule for microarray analysis. We found that 19 of these genes were approximately equally expressed, and 5 genes had significantly lower expression levels in the controls versus the ARCs as fold change >1.5 (P < 0.05).

Average relative expression of 24 DNA methylation- and transcriptional repression-associated genes in three pairs of ARCs and controls (N = 3, each #1, #2 and #16). A combination of eight genes (18S, GAPDH, GUSB, HPR1, B2 M, RPLP0, HMBS, and ACTB) was used as housekeeping genes to control the sample input. Based on controls, the 5 genes increased expression in LECs of ARCs groups (fold change >1.5,*P < 0.05)
qRT-PCR Confirmation of the mRNA Levels
qRT-PCR analysis was then conducted to validate the relative mRNA level of the 5 genes (DNMT3B, HDAC1, HDAC4, HDAC9, and MBD3) identified by microarray analysis. Figure 2 presents results of the analysis of the expression of each mRNA in LECs of the controls compared with those of the ARCs. In LECs of ARCs, all of the 5 genes were expressed at a higher level than in LECs of controls. These results were consistent with microarray data (P < 0.01).

Relative expression of mRNA levels by qRT-PCR of 20 ARCs and 20 controls LECs (#6–#12, #14, #15, and #20–# 30, respectively.),*P < 0.01
Protein Expression of the 5 Genes Differentially Expressed in LECs of Controls and ARCs
The protein expressions of DNMT3B, HDAC1, HDAC4, HDAC9, and MBD3 in LECs of controls and ARCs were detected by Western blot analysis. Figure 3 shows that lower protein levels of DNMT3B, HDACI, HDAC4, HDAC9, and MBD3 were also detected in LECs of controls than the ARCs (P < 0.01). The protein expression of five genes consisted of the qRT-PCR data.

Relative protein levels of the genes in LECs of controls and ARCs (N = 7, each#3–#5, #13, and #17–#19). a The amount of genes protein in LECs of controls and ARCs was measured by Western bolt analysis. b Relative genes protein level to GAPDH is presented as mean ± SD.*P < 0.01
Discussion
Epigenetic modifications are most commonly regulated by direct methylation of DNA and/or by posttranslational modification of histones, both of which can either repress or promote gene transcription (Suzuki and Bird 2008). Epigenetic dysregulation, promoter methylation, and silencing of DNA repair genes are implicated in ARCs (Wang et al. 2015; Li et al. 2014). In the current research, the expression profiling of DNA methylation- and transcriptional repression-associated genes have detected signals of 24 genes from the LECs of controls and ARCs. Among those 24 genes, we found 5 genes (DNMT3B, HDAC1, HDAC4, HDAC9, and MBD3) had lower expression in LECs of controls than those of ARCs. The current data indicate that those genes may be involved in epigenetic modifications in LECs of ARCs.
A study showed that DNMT3B can act as transcriptional repressors by using their ATRX domain to recruit HDAC1(Bachman et al. 2001). It has been reported that significant deacetylations at H3K9 are specifically regulated by HDAC1 (Yasui et al. 2002). Several reports also implied that DNA hypermethylation could be triggered by higher levels of histone deacetylation (Fuks et al. 2000; Robertson et al. 2000). In this study, we found that DNMT3B and HDAC1 overexpress in LECs of ARCs than those of controls. Several studies indicated that the epigenetic mechanism regulates the αA-crystalline expression in LECs of cataract (Zhou et al. 2012; Zhu et al. 2013, 2015). In our previous studies, some DNA repair genes were found to be downregulated in ARCs (Li et al. 2015; Levine et al. 2015; Palsamy et al. 2014; Li et al. 2014; Wang et al. 2015). Hypermethylation in the gene promotors in ARC group is observed when compared with the control group. The overexpression of DNMT3B and HDAC1 may be the reason for downregulation of the DNA repair genes in LECs of ARCs.
There are several mechanisms of the posttranslational modification including phosphorylation, acetylation, and ubiquitination. But there are a few researches in this field about the pathogenesis of ARC. In our previous study, OGG1 acetylation regulates its function in response to DNA damage and could be one of the mechanisms of ARC (Kang et al. 2015). Ubiquitination also effect the mutations of αA-crystallin and β-crystallin degraded (Raju and Abraham 2011; Dudek et al. 2010). HDAC4, a key member of class II HDACs, is expressed in multiple tissues. Recent evidence has demonstrated that HDAC4 plays an important role in modulation of biological responses and pathological disorders (Wang et al. 2014). Emerging evidence suggests that HDAC4 and DNMT3B are strictly linked in silencing gene expression (Gangisetty et al. 2015). A study showed that HDAC1 also interacts with HDAC9 during neuronal death (Bardai et al. 2012). HDAC9, like most class II HDACs, has a conserved histone deacetylase domain, catalyzes the removal of acetyl moieties in the N-terminal tail of histones, and possesses a long regulatory N-terminal domain to interact with tissue-specific transcription factors and corepressors (Parra and Verdin 2010). High expression of HDAC9 has been reported in several diseases (Choi et al. 2007; Bradbury et al. 2005; Milde et al. 2010). However, to our knowledge, the expression of HDAC9 in ARCs remains unclear. In this study, the higher expression of HDAC9 increased in LECs of ARCs.
Transcriptional inhibition by DNA methylation has been shown to be caused by two mechanisms, one of which is direct interference with the binding of transcriptional factors (Tate and Bird 1993) and the other recruiting of methyl-CpG-binding proteins (MBPs), which inhibit the binding of transcriptional factors to the promoter regions (Fujita et al. 1999). MBD3, a member of MBPs family, contains methyl-CpG binding domains and has a transcriptional repression function. Since most transcriptional factors do not have CpG dinucleotides within their binding sites, the silencing by DNA methylation is believed to be largely mediated by the binding of MBPs to methylated CpG dinucleotides. It has been reported that MBD3 is necessary and sufficient for the physical interaction with HDAC (Saito and Ishikawa 2002).
In summary, we demonstrated that increased expression of various epigenetic modifier genes associated with transcriptional repression. Further studies using model systems and human lens at different stages of cataract development are needed to conclude a cause–effect relationship between the gene expression changes in the lens and ARC formation. The finding in ARC might provide a proof of concept for the intervention of methylation status in ARC therapy and prevention.
References
Bachman KE, Rountree MR, Baylin SB (2001) Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J Biol Chem 276(34):32282–32287. doi:10.1074/jbc.M104661200
Bardai FH, Price V, Zaayman M, Wang L, D’Mello SR (2012) Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J Biol Chem 287(42):35444–35453. doi:10.1074/jbc.M112.394544
Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, Craddock C, Turner BM (2005) Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 19(10):1751–1759.
Choi YW, Bae SM, Kim YW, Lee HN, Park TC, Ro DY, Shin JC, Shin SJ, Seo JS, Ahn WS (2007) Gene expression profiles in squamous cell cervical carcinoma using array-based comparative genomic hybridization analysis. Int J Gynecol Cancer 17(3):687–696.
Chylack LT Jr, Wolfe JK, Singer DM, Leske MC, Bullimore MA, Bailey IL, Friend J, McCarthy D, Wu SY (1993) The lens opacities classification system III. the longitudinal study of cataract study group. Arch Ophthalmol 111(6):831–836
Dudek EJ, Lampi KJ, Lampi JA, Shang F, King J, Wang Y, Taylor A (2010) Ubiquitin proteasome pathway-mediated degradation of proteins: effects due to site-specific substrate deamidation. Invest Ophthalmol Vis Sci 51(8):4164–4173. doi:10.1167/iovs.09-4087
Fujita N, Takebayashi S, Okumura K, Kudo S, Chiba T, Saya H, Nakao M (1999) Methylation-mediated transcriptional silencing in euchromatin by methyl-CpG binding protein MBD1 isoforms. Mol Cell Biol 19(9):6415–6426
Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T (2000) DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet 24(1):88–91. doi:10.1038/71750
Gangisetty O, Wynne O, Jabbar S, Nasello C, Sarkar DK (2015) Fetal alcohol exposure reduces dopamine Receptor D2 and increases pituitary weight and prolactin production via epigenetic mechanisms. PLoS ONE 10(10):e0140699. doi:10.1371/journal.pone.0140699
Hendrich B, Bird A (1998) Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol 18(11):6538–6547
Hermann A, Gowher H, Jeltsch A (2004) Biochemistry and biology of mammalian DNA methyltransferases. Cell Mol Life Sci 61(19–20):2571–2587. doi:10.1007/s00018-004-4201-1
Hernandez-Munoz I, Taghavi P, Kuijl C, Neefjes J, van Lohuizen M (2005) Association of BMI1 with polycomb bodies is dynamic and requires PRC2/EZH2 and the maintenance DNA methyltransferase DNMT1. Mol Cell Biol 25(24):11047–11058.
Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33(Suppl):245–254. doi:10.1038/ng1089
Jin B, Robertson KD (2013) DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol 754:3–29. doi:10.1007/978-1-4419-9967-2_1
Kang L, Zhao W, Zhang G, Wu J, Guan H (2015) Acetylated 8-oxoguanine DNA glycosylase 1 and its relationship with p300 and SIRT1 in lens epithelium cells from age-related cataract. Exp Eye Res 135:102–108. doi:10.1016/j.exer.2015.02.005
Klose RJ, Bird AP (2006) Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci 31(2):89–97. doi:10.1016/j.tibs.2005.12.008
Ko YG, Nishino K, Hattori N, Arai Y, Tanaka S, Shiota K (2005) Stage-by-stage change in DNA methylation status of Dnmt1 locus during mouse early development. J Biol Chem 280(10):9627–9634.
Lei H, Oh SP, Okano M, Juttermann R, Goss KA, Jaenisch R, Li E (1996) De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development 122(10):3195–3205
Levine ME, Lu AT, Bennett DA, Horvath S (2015) Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging
Li F, Wang Y, Zhang G, Zhou J, Yang L, Guan H (2014) Expression and methylation of DNA repair genes in lens epithelium cells of age-related cataract. Mutat Res 766–767:31–36. doi:10.1016/j.mrfmmm.2014.05.010
Li W, Liu H, Yu M, Zhang X, Zhang Y, Wilson JX, Huang G (2015) Folic acid alters methylation profile of JAK-STAT and long-term depression signaling pathways in Alzheimer’s Disease models. Mol Neurobiol. doi:10.1007/s12035-015-9556-9
Ma P, de Waal E, Weaver JR, Bartolomei MS, Schultz RM (2015) A DNMT3A2-HDAC2 complex is essential for genomic imprinting and genome integrity in mouse oocytes. Cell Rep 13(8):1552–1560. doi:10.1016/j.celrep.2015.10.031
Milde T, Oehme I, Korshunov A, Kopp-Schneider A, Remke M, Northcott P, Deubzer HE, Lodrini M, Taylor MD, von Deimling A, Pfister S, Witt O (2010) HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth. Clin Cancer Res 16(12):3240–3252. doi:10.1158/1078-0432.CCR-10-0395
Nan X, Meehan RR, Bird A (1993) Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res 21(21):4886–4892
Okano M, Xie S, Li E (1998) Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 19(3):219–220. doi:10.1038/890
Palsamy P, Bidasee KR, Ayaki M, Augusteyn RC, Chan JY, Shinohara T (2014) Methylglyoxal induces endoplasmic reticulum stress and DNA demethylation in the keap1 promoter of human lens epithelial cells and age-related cataracts. Free Radic Biol Med 72:134–148. doi:10.1016/j.freeradbiomed.2014.04.010
Parra M, Verdin E (2010) Regulatory signal transduction pathways for class IIa histone deacetylases. Curr Opin Pharmacol 10(4):454–460. doi:10.1016/j.coph.2010.04.004
Raju I, Abraham EC (2011) Congenital cataract causing mutants of alphaA-crystallin/sHSP form aggregates and aggresomes degraded through ubiquitin-proteasome pathway. PLoS ONE 6(11):e28085. doi:10.1371/journal.pone.0028085
Ratnam S, Mertineit C, Ding F, Howell CY, Clarke HJ, Bestor TH, Chaillet JR, Trasler JM (2002) Dynamics of Dnmt1 methyltransferase expression and intracellular localization during oogenesis and preimplantation development. Dev Biol 245(2):304–314. doi:10.1006/dbio.2002.0628
Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP (2000) DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet 25(3):338–342. doi:10.1038/77124
Saito M, Ishikawa F (2002) The mCpG-binding domain of human MBD3 does not bind to mCpG but interacts with NuRD/Mi2 components HDAC1 and MTA2. J Biol Chem 277(38):35434–35439. doi:10.1074/jbc.M203455200
Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9(6):465–476. doi:10.1038/nrg2341
Taby R, Issa JP (2010) Cancer epigenetics. CA Cancer J Clin 60(6):376–392. doi:10.3322/caac.20085
Tate PH, Bird AP (1993) Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev 3(2):226–231
Tatematsu KI, Yamazaki T, Ishikawa F (2000) MBD2-MBD3 complex binds to hemi-methylated DNA and forms a complex containing DNMT1 at the replication foci in late S phase. Genes to cells 5(8):677–688
Vaissiere T, Sawan C, Herceg Z (2008) Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res 659(1–2):40–48. doi:10.1016/j.mrrev.2008.02.004
Wang Z, Qin G, Zhao TC (2014) HDAC4: mechanism of regulation and biological functions. Epigenomics 6(1):139–150. doi:10.2217/epi.13.73
Wang Y, Li F, Zhang G, Kang L, Qin B, Guan H (2015) Altered DNA methylation and expression profiles of 8-oxoguanine DNA glycosylase 1 in lens tissue from age-related cataract patients. Curr Eye Res 40(8):815–821. doi:10.3109/02713683.2014.957778
Yasui D, Miyano M, Cai S, Varga-Weisz P, Kohwi-Shigematsu T (2002) SATB1 targets chromatin remodelling to regulate genes over long distances. Nature 419(6907):641–645. doi:10.1038/nature01084
Zhou P, Luo Y, Liu X, Fan L, Lu Y (2012) Down-regulation and CpG island hypermethylation of CRYAA in age-related nuclear cataract. FASEB J 26(12):4897–4902. doi:10.1096/fj.12-213702
Zhu XJ, Zhou P, Zhang KK, Yang J, Luo Y, Lu Y (2013) Epigenetic regulation of alphaA-crystallin in high myopia-induced dark nuclear cataract. PLoS ONE 8(12):e81900. doi:10.1371/journal.pone.0081900
Zhu XJ, Zhang KK, Zhou P, Jiang CH, Lu Y (2015) alphaA-crystallin gene CpG islands hypermethylation in nuclear cataract after pars plana vitrectomy. Graefes Arch Clin Exp Ophthalmol 253(7):1043–1051. doi:10.1007/s00417-015-2949-7
Acknowledgments
The study was supported by the National Natural Science Foundation of China (No. 81270987).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that there are no conflicts of interest.
Rights and permissions
About this article

Cite this article
Wang, Y., Zhang, G., Kang, L. et al. Expression Profiling of DNA Methylation and Transcriptional Repression Associated Genes in Lens Epithelium Cells of Age-Related Cataract. Cell Mol Neurobiol 37, 537–543 (2017). https://doi.org/10.1007/s10571-016-0393-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-016-0393-9