
Abstract
A set of facile room temperature catalytic systems for the oxidation of cyclohexane C–H bonds was developed from in situ generated triazole-functionalised Schiff base copper complexes. The combination of a new triazolium-functionalised Schiff base, [(E)-3-methyl-1-propyl-4-(2-(((2-(pyridin-2-yl)ethyl)imino)methyl)phenyl)-1H-1,2,3-triazol-3-ium hexafluorophosphate(V), 2] with a range of bench-top Cu(I) and Cu(II) salts (Cu2O, CuO, Cu(CH3CN)4PF6, CuSO4·5H2O, Cu2(OAc)4·2H2O, CuCl2, Cu(NO3)2·3H2O) as catalysts were screened under varying reaction conditions for the peroxidation of cyclohexane using hydrogen peroxide as a green source of oxygen. High conversions to oxidised products were obtained with up to 80% in 6 h for the 2/CuSO4·5H2O system at 1 mol% catalyst concentration under optimised reaction conditions. All the copper salts yielded the ketone–alcohol (K–A) oil containing varying ratios of cyclohexanol and cyclohexanone. The results also showed that at room temperature, the various in situ generated copper catalysts exclusively yielded only the K–A oil. Furthermore, by changing the reaction temperature to reflux in acetonitrile and depending on the starting substrate (cyclohexane, cyclohexanol or cyclohexanone), 23–100% of adipic acid was also obtained. The kinetics study for the peroxidation reaction reveals activation energy of 12.29 ± 2 kJ/mol following a copper initiated radical mechanism.
Graphic Abstract

Similar content being viewed by others
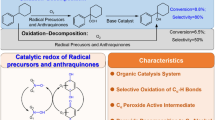

Avoid common mistakes on your manuscript.
1 Introduction
Nylons, resins, plasticisers and lubricants are produced from adipic acid (AA), which is amongst the most widely used feedstock in the textile and allied chemicals industries. At the industrial scale, AA is obtained from the nitric acid-catalysed terminal oxidation of cyclohexane (Cy-H) via its primary oxidation products, cyclohexanone (Cy=O, K) and cyclohexanol (Cy-OH, A), the so-called (ketone-alcohol) K–A oil [1, 2]. Processing the K–A oil to AA usually requires harsh conditions (i.e. high temperatures, use of strong oxidants, and stoichiometric additives), which often lead to poor selectivity, low yields, and undesired environmental and economic costs [3].
The catalytic oxidation of Cy-H to the K–A oil using green oxidants such as H2O2, tert-butyl hydroperoxide and molecular oxygen continues to be a challenge. This is because the majority of the methods for this process are based on heterogeneous catalysts. Although in comparison with homogeneous systems such as those reported herein, heterogeneous catalysts offer the advantage of catalyst reuse and recyclability, however, they are more prone to undesirable over-oxidation and the production of side products [4,5,6]. Various homogeneous copper-based catalytic systems have been developed over the years and used to catalyse Cy-H peroxidation at low temperatures with low to moderate yields [7,8,9,10,11,12,13,14,15,16,17,18,19,20]. Table 1 shows the development of homogeneous copper catalysed systems for the mild oxidation of cyclohexane to its products. The low conversions reported for many of the catalysts made their industrial application difficult, and thus far, the most active homogeneous catalysts were reported in 2016 by Garcia-Bosch and Siegler, who reported 56% conversion of the substrate (Table 1, entry 12) [18]. The compounds listed in Table 1 serve as indicators toward the development of more active and mild condition oxidation catalysts based on copper.
When compared to isolated metal complexes as catalysts, the in situ generated variants are easy to prepare, cost-effective and require minimal to no work-up procedures for their application. In situ catalysis featuring Cu/imidazolium salts was pioneered by Woodward in 2001 [21]. And since then, various Cu/imidazolium and triazolium salts were reported to be active in a wide variety of homogeneous catalytic reactions such as enantioselective C–C coupling [22, 23], α-alkylation of ketones with alcohols [24] and the oxidation of alkyl and aryl alcohols [25]. Recently we reported a set of in situ generated Cu(I)/L (L = 1,2,3-triazol-5-ylidene) catalysts that showed moderate conversions (up to 39%) for the oxidation of Cy-H (Table 1, entry 13) [19] and have previously developed a variety of azolium (imidazolium and triazolium)- based transition metal complexes for application in oxidation [26] and C-N coupling reactions [27].
Since the beginning of the twentieth century, Schiff bases have been adopted as exceptional donor ligands to stabilise reactive metal ions [28] for efficient transition metal catalysed homogenous reactions [29, 30]. Consequently, coupling a Schiff base moiety to an azolium core yields multifunctional polydentate donor ligands [31,32,33] that have shown unique and efficient stereoselectivity in asymmetric catalysis by transition metals [34]. Hence, in this report, we present the application of a new series of in situ generated copper complexes that are stabilised by such a multifunctional ligand framework (iminopyridyl-1,2,3-triazole). The three potential coordinating donor arms of the ligand will serve to stabilise the metal centres as catalysts for the peroxidation of Cy-H to its oxygenate products and, more importantly, to AA. The formation of the in situ complexes reported in this work and their application as viable catalysts for cyclohexane oxidation illustrates the potential of bench-top reagents as alternatives to more expensive conventional metal precursors in the preparation of homogeneous catalysts. Consequently, the afforded catalyst systems described herein are cheap to operate, work under mild conditions and environmentally benign [35]. This report is an effort to circumvent the shortcomings of previously reported homogeneous catalyst systems by combining the well-documented catalytic properties of the Schiff base and triazolylidene ligands with the potential hemi-lability of a pyridyl N-donor to further improve the capability of copper for oxidation catalysis.
2 Experimental
2.1 General Information
All reagents were purchased from Sigma-Aldrich and used as received. Solvents (acetonitrile, tetrahydrofuran, methanol and diethyl ether) were purchased from Merck and purified using a commercially available MBraun MB-SP Series solvent purification system equipped with activated alumina columns. Unless otherwise stated, all syntheses were performed under a dinitrogen atmosphere using standard Schlenk techniques. We recently reported the full characterization of the starting aldehyde 4-(2-formylphenyl)-3-methyl-1-propyl-1H-1,2,3-triazol-3-ium iodide, 1 [36]. NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer operated at ambient temperature with δ values reported in ppm referenced to (CH3)4Si as the internal standard for both 1H and 13C NMR data. Infrared spectra were recorded on a Perkin Elmer universal ATR Spectrum 100 FT-IR spectrometer. UV–Vis spectra were measured on a Shimadzu UV–Vis–NIR Spectrophotometer UV – 3600. Mass spectrometric data were recorded on Waters Micromass LCT Premier TOF MS-ES+.
2.2 Synthesis of the Functionalised Schiff Base Ligand Precursor Salt
(E)-3-methyl-1-propyl-4-(2-(((2-(pyridin-2-yl)ethyl)imino)methyl)phenyl)-1H-1,2,3-triazol-3-ium hexafluorophosphate(V), 2
A conventional Schiff base synthetic procedure was used [37]. A methanolic solution of a mixture of 1 (0.357 g, 1 mmol), and 2 mol equivalent of 2-(pyridin-2-yl)ethanamine (0.244 g, 2 mmol) was stirred and refluxed for 4 h in a 20 cm3 Schlenk tube. To the reaction flask was then added KPF6 (0.18 g, 1 mmol), and the mixture continuously stirred at room temperature for 12 h. Removal of the solvent followed by extraction with DCM (3 × 25 cm3) gave the crude 2 as the DCM soluble fraction. The DCM soluble fraction was then concentrated under reduced pressure, washed with diethyl ether (3 × 25 cm3) and dried to afford the pure 2, as yellow viscous oil. Yield 0.38 g, 80%. 1H NMR (DMSO-d6, 400 MHz): δ 0.96 (3H, t, J = 7.4 Hz, CH3 Pr), 1.98 (2H, m, J = 7.2 Hz, –CH2– Pr), 2.89 (2H, t, J = 14.2 Hz, –CH2–), 3.82 (2H, t, J = 6.9 Hz, –CH2–N =), 3.86 (3H, s, CH3), 4.61 (2H, t, J = 13.9 Hz, –CH2–N = pr), 7.22 (2H, t, J = 4.1 Hz, Ar), 7.60 (1H, d, J = 6.9 Hz, Ar), 7.70 (2H, t, J = 7.7 Hz, Py), 7.71 (1H, d, J = 15.3 Hz, Ar), 7.92 (1H, d, J = 7.5 Hz, Py), 8.37 (1H, s, HC = N imine), 8.48 (1H, d, J = 4.4 Hz, Py), 8.95 (1H, s, NNCH triazolyl). 13C NMR (DMSO, 100.6 MHz): δ 10.3, 22.3, 54.6, 59.9, 120.4, 121.4, 123.0, 129.1, 130.6, 131.1, 132.1, 135.1, 136.4 (NNCH triazolyl), 141.6, 148.9, 159.1, 160.1 (HC = N imine). IR (ATR, cm−1): 3051 (C–H, sp2), 1643 (C = N), 1591 (C = C), 1166 (N = N), 864 (\({\mathrm{PF}}_{6}^{-}\)), 766 (Ar C–H). HRMS (ESI +): m/z 334.2028 [(M – PF6)]+, calculated for [(M – PF6]+ 334.2032.
2.3 General Procedure for the Catalytic Oxidation Reaction
Except when mentioned, all the processes involved in the oxidation reaction were carried out in an aerobic environment in a Schlenk tube fitted with an efficient reflux condenser. Cyclopentanone was used as the internal standard, and all GC quantitative analysis and experiments were conducted with an Agilent Technology 6820 GC System equipped with a flame ionisation detector (FID) and an Agilent DB wax column with a length of 30 m, an inner diameter of 0.25 mm and a thickness of 0.25 mm. Retention times from the catalytic mixture were compared with commercial standards of cyclohexane, cyclohexanol, cyclohexanone and adipic acid. The general stepwise peroxidation of Cy-H by 2/CuSO4 is described; other catalyst systems were tested using the same procedure:
The catalyst was generated in situ from CuSO4 (19.9 mg, 40 µmols) and compound 2 (38.4 mg, 40 µmols), which were dissolved in 1 cm3 of CH3CN and placed in a 20 cm3 Schlenk tube fitted with a stirrer and an efficient reflux condenser. Then, 1.52 cm3 of H2O2 solution (30% in H2O, 20 mmol) was added to the 2/CuSO4 mixture. Immediately after that (about 10 s), then the appropriate substrate, e.g. for Cy-H (0.44 cm3, 4 mmol), was added to the mixture with the total reaction volume of 3 cm3. The reaction mixture was stirred at room temperature for peroxidation to Cy-OH and Cy=O or at reflux for the production of AA. At the end of the reaction (6 h), the internal standard cyclopentanone (0.1 cm3, 95 mg, 1 mmol) and 5 cm3 of diethyl ether were added to the reaction mixture. Then, 2 cm3 of the crude product were added to a 5 cm3 vial containing PPh3 (1.5 mmol) and placed in an ice bath in order to convert the Cy-OOH to Cy-OH (excess H2O2 is converted to H2O). After 15 min, an aliquot of the PPh3 treated sample was taken using a Pasteur pipette and filtered through a cotton wool plug. The filtrate (0.5 μL) was then injected into the GC for analysis and quantification. Products were identified by comparison with analytically genuine samples, and yields were quantified from peak areas (average of two runs within ± 5%). Treatment of the product with PPh3 is not required when Cy-OH and Cy= O were used as substrates for the production of AA [35].
3 Results and Discussion
3.1 Synthesis
We recently reported on the synthesis and full characterisation of 1 [36]. Compound 2 was synthesised from 1 by the standard Schiff-base condensation of the triazolium aldehyde and 2-(pyridin-2-yl)ethan-1-amine. The iodide salt was subjected to counterion metathesis with KPF6 to give 2, in good yield as a yellow viscous oil. All the compounds were isolated and fully characterised (Scheme 1).

Synthesis of 1,2,3-triazolium Schiff base salt, 2. Reaction conditions: a) i. 2-(pyridin-2-yl)ethan-1-amine, MeOH, 4 h; ii. KPF6, MeCN, rt, 12 h
3.2 Characterisation
Formation of salt 2 was confirmed by the disappearance of the aldehyde singlet proton signal that was initially observed in 1 at δ 9.20 ppm, and the corresponding appearance of two distinct singlet signals at 8.9 ppm and 8.4 ppm in the 1H NMR spectrum assigned to the triazolium (C5-H) and imine (HC=N) protons, respectively. The corresponding 13C NMR spectrum showed the imine (HC=N) carbon at 160 ppm and shifted upfield compared to the signal of the unreacted triazolium aldehyde that was observed at 192 ppm in 1. Aromatic and aliphatic protons of compound 2 were within the expected regions in both the 1H and 13C NMR spectra [38, 39]. The 31P NMR spectrum of compound 2 gave a multiplet peak at -148 ppm, accounting for the successful incorporation of PF6 as a counterion in 2 [27]. In addition, FTIR and HRMS complemented and unambiguously confirmed the formation of the hexafluorophosphate salt. Distinct signals corresponding to the imine (C=N) and aryl (C=C) functionalities were observed respectively at 1643 and 1591 cm−1 in the IR data of compound 2. The absence of the C=O stretching vibration band previously observed at 1702 cm−1 in 1 confirms the formation of the Schiff base functionality in 2 [39]. The pattern of the mass ionisation spectrum of 2 is similar to those reported for related compounds [40]. It is characterised by the loss of the counterion with the [(M – PF6)]+ fragment as the parent molecular ion, the HRMS analysis of which gave an m/z value that is within acceptable limits of the calculated value.
3.3 In Situ Catalytic Peroxidation of Cy-H
A study of the catalytic oxidation of Cy-H was initiated at room temperature, initially for 6 h in the preliminary optimisation stage (Scheme 2). Various parameters that included the substrate to oxidant ratio, solvent to oxidant ratio, the concentration of the catalyst and time of the reaction were studied. The precatalyst at this stage was a mixture of 2 and Cu(CH3CN)4PF6, which in solution generated the catalyst in situ. It is important to highlight that excess triphenylphosphine was added to each aliquot before injection to the GC in order to maximise the yield of Cy-OH. This has been noted to result from the reduction of the major oxidation product (Cy-OOH, Schemes 2–3) [41].

Catalytic peroxidation of Cy-H to the alcohol (Cy-OH) and ketone (Cy=O) products

Proposed reaction mechanism for the copper catalysed oxidation of Cy-H to AA
Much of the reported literature on copper catalysed oxidation catalysis (Table 1) has indicated that an appropriate ligand framework that supports the metal centre in a required state of oxidation is a prerequisite for an efficient catalytic system [42]. Hence, we carried out optimisation studies to investigate the roles of the Cu and precursor salt 2 on the Cy-H peroxidation studies. Analysis of the data obtained showed the importance of the Cu species as the active centre because no conversion of the Cy-H substrate to its oxidised products was observed in the absence of the metal. However, when the metal salt was added without the ligand precursor 2, a low substrate conversion of 8% was recorded, which is attributed to a very unstable oxidant-generated copper hydroperoxyl complex activating the substrate [42].
Then we studied the influence of varying the oxidant concentration on the catalytic process. The results presented in Fig. 1 show a direct dependence of substrate conversion on oxidant concentration with optimum efficiency at 5 mol equivalents of H2O2. In context, many catalytic systems for the peroxidation of Cy-H have reported the use of up to 10 mol equivalents of an oxidant [15, 17,18,19].

Dependence of Cy-H conversion to oxygenates on oxidant concentration
Results on the optimisation of catalyst concentration (Fig. 2) also indicated a direct correlation with substrate conversion. A high conversion of 76% was obtained at a catalyst concentration of 3 mol%. However, the best catalyst efficiency as turnover number (TON) was obtained at a catalyst concentration of 0.25 mol%, albeit at a low substrate conversion of 23% (Fig. 2).

Catalyst efficiency as a function of concentration
An exploration of previous reports on this subject has shown that catalyst concentrations above 1 mol% were associated with slower reactions, decreased conversions supplemented by low turnover values, all of which were attributed to the decomposition of available active sites at higher concentrations of a catalyst [15, 18, 19, 43]. In summary, a precatalyst concentration of 1 mol% was adopted for the remainder of this study.
A study on the reaction time profile presented in Fig. 3 shows that five molar equivalents of the oxidant and 1 mol% of the in situ generated catalyst led to 91% conversion in 24 h. However, evaluation of the catalyst efficiency as turnover frequency (TOF) indicates that the highest TOF of 103 h−1 was achieved at 6 h; hence the remainder of the study was conducted using the optimised conditions of 1 mol% catalyst and 5 mol% H2O2 at 6 h reaction time.

Time profile for the peroxidative conversion of Cy-H
Metal catalysed peroxidation reactions have been reported to proceed through the formation of radical species or Fenton-type routes. Thus, we added a stoichiometric amount of a radical scavenger (PH2NH) at the start of the reaction as an inhibitor to gain some insight into the route involved in the peroxidative catalysis [35, 44]. This led to only 4% conversion of Cy-H to the oxygenate products, which is an indication that the radical scavenger has shut down the catalysis, clearly suggesting the active participation of radical species in the reaction [14].
A revision of previously reported literature indicates that ligand stabilised metal centres serve as good catalysts in reactions that proceeded via Fenton-type mechanisms, with the supporting ligand serving to mediate the extent of metal-substrate interactions [44]. In these regards, we recently reported a monodentate triazolylidene copper complex that actively yielded 39% conversion of Cy-H to a mixed ketone-alcohol product [19]. Thus, we are of the opinion that the multidentate ligand framework presented in this study in conjunction with copper will lead to even better catalytic activity due to enhanced ligand/metal interaction and cooperation. This assertion is further supported by our recent report involving Cu(I) compounds as catalysts [45]. Hence, we proceeded to study the efficiency of a range of bench-top Cu(I) and Cu(II) salts combined with the triazolium Schiff base ligand precursor 2 to generate in situ catalysts with the results presented in Fig. 4. Interestingly, all the metal salts gave good to excellent overall conversions in the range of 51–80%. The Cu(II) salts accounted for higher conversions to the oxygenate products, with CuSO4 yielding the best conversion of 80%. All the salts yielded the K-A oil in varying combinations, with the ketones being dominant for the majority of the salts that are Cu(II) centered. This general trend agrees with other literature reports where Cy=O was the major product of Cy-H peroxidation reaction [7,8,9,10,11,12,13,14,15,16,17]. It is interesting to note that both Cu(I) and Cu(II) oxides and the other Cu(I) salts preferentially produced the alcohol in higher quantities, indicating that these salts produced more of the major product (CyOOH) and curtailed secondary oxidation to the ketone. However, in practical terms, this will minimally affect the further oxidation of the K-A oils from these salts to the terminal (AA) product (see Scheme 2).

Catalytic performance of a range of bench-top Cu(I) and Cu(II) salts for the oxidation of Cy-H to the K-A oil
3.4 Catalytic Peroxidation of Cy-H to AA
Previously reported studies on Cu-containing catalyst systems geared toward the oxidation of Cy-H were limited to optimising the conversion of the substrate to the primary oxygenate (K-A) product (Table 1), which in its own right is a very important feedstock for the industrial production of nylons and related products [11, 42]. In this study (Table 2), we present a viable and environmentally benign system for the direct quantitative production of AA, a very important raw material for many industrial chemical processes. The process involves the terminal oxidation of Cy-H to AA under mild reaction conditions, such that instead of the room temperature reactions studied thus far, refluxing neat Cy-H (70 °C) under the optimised reaction conditions (catalyst, oxidant, time) did not only produce higher conversions (up to 96%) but, more significantly, directly yielded 23% of AA. This is contrary to the room temperature studies in which AA was not observed at any stage of the reaction. Furthermore, using Cy-OH as the starting substrate under reflux conditions gave about 80% total conversion with a much-improved selectivity (55%) to AA. Even more impressive is the direct oxidation of Cy=O that exclusively yielded 100% AA, albeit at a low 21% conversion. It is clear from this set of results that the peroxidation of Cy-H or its main products (K-A) at an elevated reflux temperature has resulted in the production of quantifiable quantities of the terminal adipic acid product. The reflux condition supplied the required energy input to the catalytic system for overcoming the energy barriers of Cy-H ring scission via C–C bond cleavage for the production of the AA product (Scheme 2).

In order to contextualise the current study, Table 2 presents a comparative analysis on the activities of related homogeneous catalysts reported in the literature for the peroxidation of Cy-H to oxygenated products, including AA. It is important to note that there is a paucity of reports for the homogeneous mild condition direct production of AA from Cy-H (or its primary products, Cy-OH and Cy=O) [35, 44, 46, 47]. The use of metal-based precatalysts and oxygen as the oxidant has dominated the reports in the literature with 12–73% selectivity to AA (Table 2, entries 4–7). Another interesting homogeneous system (Table 2, entry 8) that involves the use of N-hydroxyphthalimide as an organocatalyst required a harsh oxidation environment (HNO3/trifluoroacetic acid) to selectively yield 82% AA from Cy-H. This catalytic system resembles the industrial conversion of KA oil to AA, only that in this case, a metal catalyst is not required, which is an advantage for end-uses where extreme high purity of the product is required. A current study by Becker and co-workers, which is the most closely related to this study (Table 2, entry 9), in which H2O2 was used as an oxidant with a Cu cluster as the catalyst under very mild conditions produced what is to date the best mild reaction conversion of Cy-H at 95% with an impressive 45% selectivity to AA. In comparison, the 2/Cu in situ catalyst system presented herein (Table 2, entry 1) has closely mirrored the efficiency of the Cu cluster system. In fact, we report here that when the starting substrate is Cy-OH, the selectivity to AA is improved to 55% (Table 2, entry 2) and up to 100% (Table 2, entry 3) when Cy=O was used as the substrate. Hence, we opine that the 2/Cu system is a very robust, simpler and highly efficient system for the production of AA from components of the KA oil based on a bench-top Cu salt that is comparable to the best systems reported to date.
3.5 Kinetic Studies
Kinetic parameters were determined using the method of initial rates, whereby in all the experiments, the rate of Cy-H conversion to the oxygenate products was monitored by GC. Firstly, the initial quantities of the oxidant, Cy-H, solvent and temperature were kept constant, and the concentration of the in situ generated precatalyst was varied (Fig. 5A). The slope of the graph was used to determine the initial rate constants. Plots of the rates vs concentration of the precatalyst (Fig. 5B) gave a linear relationship consistent with a first-order dependence.

Influence of catalyst [2/CuSO4·5H2O] concentration on Cy-H conversion (A); determination of initial rate thereof (B). Reaction conditions: Room temperature; [Cy-H] = 0.4 mmol; [H2O2] = 2 mmol; total reaction volume [in acetonitrile] = 3 cm3
Similarly, the kinetic order with respect to the oxidant was determined by varying the concentration of H2O2 only, and the results presented in Fig. 6 show a linear increase in the initial reaction rate with an increase in oxidant concentration, thereby signifying first-order dependence as well. However, initial rates determination with respect to the substrate shows that a variation of the concentration of Cy-H (Fig. 7) had no influence, and the kinetic order of the reaction was observed to remain constant, indicating a zero-order dependence. The nature of the dependence of reaction kinetics on experimental factors (catalyst, oxidant and Cy-H concentrations) recorded herein seems standard and in agreement with recent reports on a series of copper(I) catalysts bearing a variety of N-donor ligands that were also utilised for the oxidation of Cy-H [13, 18].

Influence of changes in oxidant [H2O2] concentration on Cy-H conversion (A); determination of initial rate thereof (B). Reaction conditions: room temperature; [Cy-H] = 0.4 mmol; [2/CuSO4.5H2O] = 0.04 mmol; total reaction volume [in acetonitrile] = 3 cm3

Influence of variation in substrate concentration on Cy-H conversion (A); determination of initial rate thereof (B). Reaction conditions: room temperature; [H2O2] = 2 mmol; [2/CuSO4.5H2O] = 0.04 mmol; total reaction volume [in acetonitrile] = 3 cm3
The activation energy (Ea) was determined from an Arrhenius plot (Fig. 8) as 12.29 ± 2 kJ/mol, which is substantially lower than the average bond dissociation energy of a standard C-H bond (416.11 kJ/mol) [48]. We attribute the substantial lowering of the energy barrier required for the activation of the cyclohexane C-H bond to the catalytic conditions developed for the oxidation reaction, which points to a catalyst moderation of the energy input needed for activation even though the reactions were conducted at mild room temperature conditions or reflux for AA [49]. We believe that a metal–ligand cooperative effect is mainly responsible for the substantial conversions of the substrates recorded (see below) [50].

Arrhenius plot for the determination of oxidation reaction activation energy (Ea)
3.6 UV–Vis Spectral Study for the Determination of Active Cu Species
A common observation that we noted whilst generating and testing these series of catalytic systems is the vigorously exothermic nature and visible colour changes that occurred during the initial stages of each reaction run, usually from colourless to yellow and finally, light green (depending on the source of copper) colour. These changes are attributed to ligand coordination to Cu and the eventual generation of a Cu(II) hydroperoxy complex, a key intermediate in the oxidation reaction cycle. This is supported by a UV study conducted on the ligand precursor salt 2, and the corresponding in situ generated copper complexes (Fig. 9). The UV spectrum of the ligand precursor salt (Fig. 9, black) is featureless and only showed a shoulder band at 282 nm assigned to the π–π* transition of the pyridyl and phenyl rings [51, 52]. However, on the addition of the copper salt (Fig. 9, red), a new shoulder tail appeared at 364 nm, which we attribute to the binding of a ligand to the metal (via the imine C = N–) and in agreement with reported literature [52, 53], it is assigned to the n-π* transition of Cu(I)–N bond. Interestingly, the addition of 0.2 cm3 of the 30% H2O2 solution (1 mM) to the in situ generated complex solution led to the emergence of a new band at 356 nm (Fig. 9, green) assigned to the n-π* transition of copper hydroxide (Cu(II)-OH) complex [52]. This new band only lasts for about 5 min (Fig. 9; blue), and after 10 min (Fig. 9; cyan), it has reduced to a shoulder tail which continued to decline in intensity for 40 min (Fig. 9; brown) before merging with the 364 nm shoulder tail of the starting copper complex. The addition of a further 0.2 cm3 of fresh hydrogen peroxide led to an increase in its intensity (Fig. 9; dark purple), a piece of clear evidence for the formation of Cu(II)-OH complex, which is the active species that generate the hydroxyl radical responsible for activating the substrate (Scheme 3). The role of the ligand precursor 2 is to stabilise this copper complex, which was observed by UV for over 40 min after the initial addition of the hydrogen peroxide. However, on the addition of Cy-H to the reaction mixture, no change in the UV spectrum was observed even after 6 h. Likewise, Garcia-Bosch and Siegler also confirmed the generation of copper peroxo species using UV–visible analysis [18].

UV–Visible spectroscopic analysis of the peroxidation of Cy-H catalysed by 2/Cu(CH3CN)4PF6 using H2O2 as oxidant
3.7 Proposed Reaction Mechanism
As noted earlier, kinetics studies indicated the dependence of the reaction rate on the catalyst and oxidant concentrations whilst the addition of a radical scavenger Ph2NH was observed to shut down the oxidation reaction. Hence, a copper initiated radical mechanism is proposed in line with our report for related copper(I) complexes for the formation of the K-A oil as the main product [19]. The proposed mechanism (Scheme 3) involves a reversible one-electron redox process of the in situ generated copper catalyst [Cu(I) or Cu(II)]. The catalyst is first oxidised to generate the active Cu-OH species and a hydroxyl (HO.) radical (i). This radical was consumed in the presence of Ph2NH, as noted above. The HO. radical then activates the substrate (Cy-H) by abstracting a proton leading to the formation of a cyclohexyl radical (Cy., ii), which further reacts with available oxygen from the oxidant to form the cyclohexylperoxyl radical (Cy-OO., iii). The most crucial step is the reduction of Cy-OO. to cyclohexyl hydroperoxide (CyOOH) and regenerating Cy. (iv). This is because the alkyl hydroperoxide species has been identified as the primary product in alkane peroxidation reactions. In order to maximise product formation, the standard practice is to reduce the CyOOH to cyclohexanol (Cy-OH) using a suitable reductant, often PPh3 (v). This procedure developed by Shulpin is well-recognised for the oxidation of alkanes [54]. In addition, due to the higher reactivity of Cy-OH relative to the substrate Cy-H, many peroxide catalysed reactions have reported the ketone (Cy=O, vi) as the primary product (Table 1) which concurs with the results presented in Fig. 4.
In the second part of the reaction involving refluxing catalytic conditions, Cy-OH and Cy=O are converted to the terminal product (AA) following a series of sequential oxidation and hydrolysis reactions [55,56,57,58]. The true nature of the intermediary products (vii–ix) is still a subject of debate; however, based on the final products observed by GC in this study, a plausible route is presented in Scheme 3. The route is an adaptation from a report by Usui and Sato, who also noted their inability to observe any of the elusive intermediates (vii–ix) [57].
4 Conclusions
A simple strategy for the production of adipic acid from cyclohexane and its immediate oxidation products (cyclohexanol and cyclohexanone) was developed and presented. The key finding of this study is the use of refluxing conditions with bench-top copper salts and a new multifunctional imino-triazolium ligand precursor salt to generate in situ catalysts for the oxidation reaction promoted by a green oxidant, 30% H2O2 in water. When benchmarked against previously reported homogeneous copper catalysts for cycloalkane oxidation reactions, the current method is a novelty for homogeneous systems for adipic acid production. In addition, this method has the potential to be developed into a viable route for the large-scale production of a very desirable and large volume industrial feedstock that is a raw material for a wide range of commercial applications, notably for the production of nylon polymers. A brief insight into the kinetics and mechanism of the oxidation process indicated a radical catalytic process that is catalysed by the in situ generated copper catalysts.
References
Schuchardt U, Cardoso D, Sercheli R et al (2001) Cyclohexane oxidation continues to be a challenge. Appl Catal A 211:1
Castellan A, Bart JCJ, Cavallaro S (1991) Industrial production and use of adipic acid. Catal Today 9:231
Musser MT (2000) Ullmann’s encyclopedia of industrial chemistry. Wiley, Weinheim
Anumula R, Cui C, Yang M et al (2019) Catalytic oxidation of cyclohexane on small silver clusters supported by graphene oxide. J Phys Chem C 123:21504
Li Y, Liu C, Yang W (2017) Synthesis of porous polymeric metalloporphyrins for highly efficient oxidation of cyclohexane in heterogeneous systems. New J Chem 41:8214
Zhou L, Xu J, Miao H et al (2005) Catalytic oxidation of cyclohexane to cyclohexanol and cyclohexanone over Co3O4 nanocrystals with molecular oxygen. Appl Catal A 292:223
Derek HRB, Stephane DB, Warinthorn C et al. (1992) The functionalisation of saturated hydrocarbons. Part XXI. The Fe(III)-catalysed and the Cu(II)-catalysed oxidation of saturated hydrocarbons by hydrogen peroxide: a comparative study. Tetrahedron 48(14):2895
Andrzej S, Aimin QXL, Antoni L, Sawyer DT (1993) Copper(I)/(t-BuO0H)-induced activation of dioxygen for the ketonization of methylenic carbons. J Am Chem Soc 115(2):609
Shun-Ichi M, Naruyoshi K, Yukiko H, Kumano T (2001) Copper complexes for catalytic, aerobic oxidation of hydrocarbons. Pure Appl Chem 73(2):311
Takashi O, Ohba S, Nishida Y (1997) Oxidation of cyclohexane with hydrogen peroxide catalysed by copper(ll) complexes containing N, N-bis(2-pyridylmethyl)-B-alanineamide ligands. Polyhedron 16(21):3765
Ulf S, Ricardo P, Rufo M (1998) Iron (III) and copper (II) catalysed cyclohexane oxidation by molecular oxygen in the presence of tert-butyl hydroperoxide. J Mol Catal A 135:257
Velusamy S, Punniyamurthy T (2003) Copper(II) catalysed C-H oxidation of alkylbenzenes and cyclohexane with hydrogen peroxide. Tetrahedron Lett 44(50):8955
Shul’pin GB, Gradinaru J, Kozlov YN (2003) Alkane hydroperoxidation with peroxides catalysed by copper complexes. Org Biomol Chem 1:3611
Kirillov AM, Kopylovich MN, Kirillova MV et al (2005) Multinuclear copper triethanolamine complexes as selective catalysts for the peroxidative oxidation of alkanes under mild conditions. Angew Chem 44(28):4345
Alexander MK, Maximilian NK, Marina VK et al (2006) Mild peroxidative oxidation of cyclohexane catalysed by mono-, di-, tri-, tetra- and polynuclear copper triethanolamine complexes. Adv Synth Catal 348:159
Shimokawa C, Teraoka J, Tachi Y, Itoh S (2006) A functional model for pMMO (particulate methane monooxygenase): hydroxylation of alkanes with H2O2 catalysed by beta-diketiminatocopper(II) complexes. J Inorg Biochem 100(5–6):1118
Conde A, Vilella L, Balcells D et al (2013) Introducing copper as catalyst for oxidative alkane dehydrogenation. J Am Chem Soc 135(10):3887
Garcia-Bosch I, Siegler MA (2016) Copper-catalysed oxidation of alkanes with H2O2 under a Fenton-like Regime. Angew Chem Int Ed 55:12873
Mncube SG, Bala MD (2019) Homogeneous oxidation reactions catalysed by in situ-generated triazolylidene copper(I) complexes. Transit Met Chem 44:145
Schuchardt U, Cavalho WE, Spinacé EV (1993) Why is it interesting to study cyclohexane oxidation? Synlett 10:713
Paul KF, Woodward S (2001) Strong ligand accelerated catalysis by an Arduengo-type carbene in copper-catalysed conjugate addition. Tetrahedron Lett 42:2747
Aikomari G, Hoveyda AH (2010) Enantioselective synthesis of allylboronates bearing a tertiary or quaternary b-substituted stereogenic carbon by NHC-Cu-catalyzed substitution reactions. J Am Chem Soc 132:10634
Shintani R, Takatsu K, Takeda M, Hayashi T (2011) Copper-catalysed asymmetric allylic substitution of allyl phosphates with aryl- and alkenylboronates. Angew Chem 50(37):8656
Tan DW, Li HX, Zhang MJ et al (2017) Acceptorless dehydrogenation of alcohols catalysed by Cu(I) N-heterocycle thiolate complexes. ChemCatChem 9(6):1113
Liu X, Xia Q, Zhang Y et al (2013) Cu-NHC-TEMPO catalysed aerobic oxidation of primary alcohols to aldehydes. J Org Chem 78(17):8531
Mncube SG, Bala MD (2019) Symmetric triazolylidene Ni(II) complexes applied as oxidation catalysts. Polyhedron 157:467
Ibrahim H, Bala MD (2015) Air stable pincer (CNC) N-heterocyclic carbene–cobalt complexes and their application as catalysts for C-N coupling reactions. J Organomet Chem 794:301
Kuchtanin V, Kleščíková L, Šoral M et al (2016) Nickel(II) Schiff base complexes: synthesis, characterisation and catalytic activity in Kumada-Corriu cross-coupling reactions. Polyhedron 117:90
Fu D, Yaguang S, Stijn M et al (2008) Ruthenium complexes containing bidentate Schiff base ligands as precursors of homogeneous and immobilised catalysts. Curr Org Synth 5:291
Wesley JA, Kalidasa MK, Neelakantan MA (2014) Review on Schiff bases and their metal complexes as organic photovoltaic materials. Renew Sust Energ Rev 36:220
Abubakar S, Ibrahim H, Bala MD (2019) Transfer hydrogenation of ketones catalysed by a trinuclear Ni(II) complex of a Schiff base functionalised N-heterocyclic carbene ligand. Inorganica Chim Acta 484:276
Al-Thagfi J, Dastgir S, Lough AJ, Lavoie GG (2010) Synthesis and structural characterisation of the first copper(I) complexes with bis(imino)-N-heterocyclic carbene NCN pincer ligands. Organometallics 29(14):3133
Al-Thagfi J, Lavoie GG (2012) Synthesis, characterisation, and ethylene polymerisation studies of chromium, iron, and cobalt complexes containing 1,3-bis(imino)-N-heterocyclic carbene ligands. Organometallics 31(6):2463
Pye DR, Mankad NP (2017) Bimetallic catalysis for C-C and C-X coupling reactions. Chem Sci 8(3):1705
Matsumoto Y, Kuriyama M, Yamamoto K et al (2018) Metal-free synthesis of adipic acid via organocatalytic direct oxidation of cyclohexane under ambient temperature and pressure. Org Process Res Dev 22:1312
Lawal NS, Bala MD (2020) Click synthesis and characterisation of 1,2,3-triazolium salts. J Mol Struct 1200:127124
Cozzi PG (2004) Metal-Salen Schiff base complexes in catalysis: practical aspects. Chem Soc Rev 33(7):410
Schmid J, Frey W, Peters R (2017) Polynuclear enantiopure salen–mesoionic carbene hybrid complexes. Organometallics 36(21):4313
Grivani G, Khalaji AD, Tahmasebi V et al (2012) Synthesis, characterisation and crystal structures of new bidentate Schiff base ligand and its vanadium(IV) complex: the catalytic activity of vanadyl complex in epoxidation of alkenes. Polyhedron 31(1):265
Ibrahim H, Bala MD (2016) Earth abundant metal complexes of donor functionalised N-heterocyclic carbene ligands: synthesis, characterisation and application as amination catalysts. New J Chem 40(8):6986
Shul’pin GB, Camilla CG, Georg S et al (2005) Alkane oxygenation with H2O2 catalysed by FeCl3 and 2,2-bipyridine. Tetrahedron Lett 46(27):4563
McCann SD, Stahl SS (2015) Copper-catalysed aerobic oxidations of organic molecules: pathways for two-electron oxidation with a four-electron oxidant and a one-electron redox-active catalyst. Acc Chem Res 48(6):1756
Derek HRB, Dario D, Geletil YV (1991) The efficient oxidation of alkanes by hydrogen peroxide in pyridine mixed solvents catalysed by copper and other transition metal salts. Mendeleev Commun 1(3):115
Gawlig C, Schindler S, Becker S (2020) One-pot conversion of cyclohexane to adipic acid using a µ4-oxido-copper cluster as catalyst together with hydrogen peroxide. Eur J Inorg Chem 248
Lawal NS, Ibrahim H, Bala MD (2021) Cu(I) mediated hydrogen borrowing strategy for the α-alkylation of aryl ketones with aryl alcohols. Monatsh Chem 152(2):275
Iwahama T, Syojyo K, Sakaguchi S, Ishii Y (1998) Direct conversion of cyclohexane into adipic acid with molecular oxygen catalysed N-hydroxyphthalimide combined with Mn(acac)2 and Co(OAc)2. Org Process Res Dev 2:255
Hu B-Y, Yuan Y-J, Xiao J et al (2008) Rational oxidation of cyclohexane to cyclohexanol, cyclohexanone and adipic acid with air over metalloporphyrin and cobalt salt. J Porphyrins Phthalocyanines 12:27
Xue XS, Ji P, Zhou B, Cheng JP (2017) The essential role of bond energetics in C-H activation/functionalisation. Chem Rev 117(13):8622
Sylvain G, Catherine SJC, Nolan SP (2012) N-heterocyclic carbene gold(I) and copper(I) complexes in C-H bond activation. Acc Chem Res 45(6):778
Tan CP, Che Man YB, Selamat J, Yusoff MSA (2001) Application of arrhenius kinetics to evaluate oxidative stability in vegetable oils by isothermal differential scanning calorimetry. J Am Oil Chem Soc 78(11):1133
Walaa HM, Gehad GM, El-Dessouky MMI (2014) Synthesis, characterisation and in vitro biological activity of mixed transition metal complexes of lornoxicam with 1,10-phenanthroline. Int J Electrochem Sci 9:1415
Sun YX, Gao XH (2011) Synthesis, characterisation, and crystal structure of a new Cu(II) complex with salen-type ligand. Synth React Inorg Met-Org Nano-Met Chem 41(8):973
Benramdane R, Benghanem F, Ourari A et al (2015) Synthesis and characterisation of a new Schiff base derived from 2,3-diaminopyridine and 5-methoxysalicylaldehyde and its Ni(II), Cu(II) and Zn(II) complexes. Electrochemical and electrocatalytical studies. J Coord Chem 68(3):560
Shulpin GB (2002) Metal-catalysed hydrocarbon oxygenations in solutions: the dramatic role of additives: a review. J Mol Catal A 189:39
Chen L, Zhou T, Chen L et al (2011) Selective oxidation of cyclohexanol to cyclohexanone in the ionic liquid 1-octyl-3-methylimidazolium chloride. Chem Commun 47(33):9354
Wang L, Chen Z, Huang M et al (2016) A green route to cyclohexanone: selective oxidation of cyclohexanol promoted by non-precious catalyst of h-WO3 nanorods. Catal Lett 146(7):1283
Usui Y, Sato K (2003) A green method of adipic acid synthesis: organic solvent- and halide-free oxidation of cycloalkanones with 30% hydrogen peroxide. Green Chem 5(4):373
Zhang S-G, Jiang H, Gong H, Sun Z-l (2003) Green catalytic oxidation of cyclohexanone to adipic acid. Pet Sci Technol 21(1–2):275
Acknowledgements
We acknowledge financial support from the University of KwaZulu-Natal, ESKOM (TESP2019) and the NRF. NSL thanks Ahmadu Bello University for a paid study fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lawal, N.S., Ibrahim, H. & Bala, M.D. Facile Peroxidation of Cyclohexane Catalysed by In Situ Generated Triazole-Functionalised Schiff Base Copper Complexes. Catal Lett 152, 1264–1275 (2022). https://doi.org/10.1007/s10562-021-03732-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-021-03732-3