
Abstract
CeO2-supported LaMnO3 perovskite oxides were prepared to study their catalytic properties in the oxidation of NO to NO2. To prepare the catalyst and investigate the interaction between LaMnO3 and CeO2, two deposition methods were used. Extended X-ray absorption fine structure studies confirmed that perovskite oxide phases were formed on the CeO2 support. Moreover, X-ray photoelectron spectroscopy and temperature-programmed reduction with H2 studies revealed that the reduction temperatures for perovskite oxides and CeO2 support decreased by the deposition followed by calcination at 650 °C, and that the interaction between the LaMnO3 and CeO2 support can be controlled by changing the preparation method. The LaMnO3/CeO2 catalyst in which LaMnO3 was highly dispersed on CeO2 exhibited higher NO oxidation activity than either LaMnO3 or CeO2. The thermal stability of the LaMnO3/CeO2 catalyst was compared with that of alumina-supported LaMnO3 catalysts over 850–1050 °C.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Automobile emission control technologies are attracting considerable attention due to the severe tightening of regulations and increasing vehicle production volumes. Catalytic NO oxidation is a critical process for the purification of diesel exhaust because the resulting NO2 species is a key substance that promotes the oxidation of diesel soot and facilitates selective catalytic reduction and NOx storage under lean burn conditions. Supported Pt catalysts exhibit high activity in the oxidation of NO to NO2 [1–3]. However, it is essential that the amount of noble metals used in the catalysts be reduced or replaced by less expensive materials.
Perovskite-type mixed oxides, represented by a ABO3-type formula, have been widely used as catalysts for oxidation processes [4–6]. Perovskite oxides are good substitute candidates for noble metal catalysts [7]. Perovskite oxides containing Mn [8–10] and Co [8, 11, 12] at the B site and La at the A site have been frequently used for the catalytic oxidation of NO to NO2. In perovskite oxides, the A-site cations are partially substituted by Sr [13, 14] and Ce [15] and the B-site cations are partially substituted by other transition metals [16, 17]. Thus, one method for enhancing the catalytic activity of perovskite oxides is to substitute constituent metals and cations. Another method is to deposit them on supporting materials with a high surface area. To date, ZrO2 [18, 19], Al2O3 [20], CeO2 [21], and SiO2-base mesoporous materials [22, 23] have been used to support perovskite deposition. The supported perovskite oxides reportedly exhibit high catalytic activity and thermal stability, which effectively offset the drawback of perovskite oxides—their low surface area.
We recently reported the selective deposition of the perovskite oxides LaFeO3 and LaMnO3 inside or outside the pore of γ-Al2O3 [24, 25]. These supported perovskite oxide catalysts exhibited higher activity and thermal stability than an alumina-supported Pt catalyst when the perovskite phases were deposited inside the alumina pore. These findings inspired us to investigate the effect of supporting materials on the catalytic properties of supported perovskite oxides. Here, we investigated the effect of depositing LaMnO3 perovskite oxides onto a CeO2 support on the catalytic properties of the oxidation of NO to NO2. Because CeO2 itself is active with respect to oxidation reactions, we anticipated that the combination of CeO2 with perovskite oxide would improve the catalytic properties of the supported catalysts. We also investigated the effect of the deposition method on their catalytic properties and thermal stabilities, which were important factors when the catalysts were put into practical use for automobile emission control.
2 Experimental
2.1 Materials
The catalyst support CeO2 (JRC-CEO-3; SBET = 72 m2 g−1) was obtained from the Catalysis Society of Japan and was used after calcination at 650 °C for 5 h in air. γ-Al2O3 (JRC-ALO-8) was also used as a supporting material after the same treatment.
2.2 Preparation of LaMnO3/CeO2 Catalysts
Bulk LaMnO3 perovskite oxides were prepared using the hydrolysis-precipitation method described in a previous paper [26]. The aqueous solution containing La(NO3)3 (Wako Pure Chem) and Mn(NO3)2·6H2O (Wako Pure Chem) were slowly added to aqueous ammonia while vigorously stirring. The resulting precipitates were calcined at 650 °C for 5 h in air. The LaMnO3 catalyst thus prepared has orthorhombic structure with the tolerance factor of 0.954. For the preparation of physical mixture of LaMnO3 and CeO2, 0.20 g of LaMnO3 and 0.80 g of CeO2 were ground with a mortar and pestle. The sample was denoted by LaMnO3-MM.
To prepare CeO2-supported LaMnO3 (LaMnO3/CeO2) catalysts by the dry impregnation (DI) method, CeO2 was impregnated with a mixed aqueous solution of metal nitrates in a quantity equal to 50–75 % pore volume of the CeO2, followed by drying at 100 °C. The impregnation process was repeated until LaMnO3 loading was increased to 20 wt%. The obtained samples were then calcined at 650–1050 °C for 5 h in air. The LaMnO3/CeO2 catalyst prepared by the DI method was denoted as LaMnO3/CeO2-DI.
LaMnO3/CeO2 catalysts were also prepared by a precipitation–deposition (PD) method. Hydroxide precursors of perovskite oxide were obtained by the hydrolysis–precipitation method described above. They were then mixed well in distilled water (300 mL) with CeO2, followed by ultrasonication. Next, they were evaporated to dryness under vigorous stirring followed by grinding and calcination at 650 °C for 5 h in air. In this process, the loading level of perovskite phase after calcination depends on the water content of the hydroxide precursor. For the determination of LaMnO3 loading on the CeO2 support, therefore, the water content of the hydroxide precursor was measured by thermogravimetry (Shimadzu DTG-60) prior to the impregnation of the hydroxide precursor. The LaMnO3/CeO2 catalyst prepared by the PD method was denoted as LaMnO3/CeO2-PD.
2.3 Catalyst Characterization
The crystal structure of the catalysts was examined by X-ray diffraction (Rigaku Ultima IV) using CuKα radiation. The catalyst surface area was determined using Brunauer–Emmett–Teller (BET) plots obtained from the N2 adsorption isotherms at 77 K (Quantachrome NOVA2000).
La K-edge extended X-ray absorption fine structure (EXAFS) spectra were taken with the photon factory advanced ring at the High Energy Accelerator Research Organization (NW-10A), with the storage ring operating at an energy of 6.5 GeV. Fourier transform-EXAFS spectra were obtained from k3-weighted EXAFS data [k3χ(k)] at 3.0–12.0 Å−1. The coordination numbers (CNs), bond distances (R), Debye–Waller factor (σ2), and energy shift (∆E) were obtained using the theoretical backscattering amplitude and phase shift functions calculated by the program FEFF8 [27].
X-ray photoelectron spectroscopy (XPS) spectra were recorded using a Kratos AXIS-165 spectrometer with an Al Kα source. The binding energies were corrected using the value of 284.8 eV as an internal standard for the C 1s peak of the carbon species on the catalyst samples.
Temperature-programmed reduction with H2 (H2-TPR) was conducted with a BELCAT-30 catalyst analyzer (BEL JAPAN, Inc). The sample (0.050 g) was pretreated in an O2 flow for 2 h at 823 K. In the H2-TPR measurements, catalyst samples were heated at 10 °C/min.
2.4 Catalytic Activity Measurement
The catalytic oxidation of NO was performed using a fixed bed flow reaction system. Catalyst samples meshed at 250–750 µm were placed in the U-shaped glass reactor, which was connected to the system. The reaction gas, with a NO 500 ppm-O2 5 %-N2 balance composition, was fed to the reactor at 200–500 °C. In a typical reaction, the catalyst weighed 0.10 g and the gas flow was 150 mL/min (W/F = 0.04 g s mL−1). The concentrations of NO and NO2 were determined using a NOx analyzer (SHIMAZU NOA-7000). Prior to the reaction, the catalyst was heated in an O2 flow at 550 °C. The oxidation of NO to NO2 consists of both forward reaction (NO + 1/2O2 → NO2) and backward reaction (NO2 → NO + 1/2O2). Therefore, the steady-state activity was dominated by reaction equilibrium. The equilibrium curve for conversion of NO to NO2 was calculated based on the van’t Hoff Eqs. (1) and (2), where KP, ∆H, R, T, \({{\text{p}}_{\text{NO}}}\), \({{\text{p}}_{\text{N}{{\text{O}}_{2}}}}\), and \({{\text{p}}_{{{\text{O}}_{2}}}}\)were equilibrium constant expressed in terms of partial pressures of gases, the enthalpy of reaction, gas constant, reaction temperature, partial pressures of NO, NO2, and O2.
The reaction rate was obtained under conditions in which the NO conversion was linear with respect to the ratio of catalyst weight to gas flow rate.
3 Results and Discussion
3.1 Structure of Catalysts
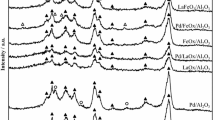
Figure 1 shows the XRD patterns of the CeO2, LaMnO3, and LaMnO3/CeO2 catalysts. The patterns of the CeO2 support and LaMnO3/CeO2 combined were almost the same as those for the CeO2 support alone, indicating that the structures of the CeO2 support were unchanged after deposition and the post-heating treatment. The peaks due to the perovskite were detected for the physical mixture of LaMnO3 and CeO2 (LaMnO3-MM) and the LaMnO3/CeO2 catalysts prepared by the PD. In contrast, perovskite phase was hardly detected for the LaMnO3/CeO2 catalyst prepared by the DI method, indicating that the LaMnO3 perovskite phases were highly dispersed on the CeO2 support.

XRD patterns of LaMnO3 and LaMnO3/CeO2 catalysts prepared by different methods
The BET surface areas of the LaMnO3/CeO2 catalysts are listed in Table 1. The surface area of LaMnO3/CeO2 catalysts was lower than that of the CeO2 support when the catalyst was prepared by the DI method, although the surface area of LaMnO3/CeO2 catalysts was much higher than that of the unsupported LaMnO3 catalysts. It is noted that the surface area of the catalyst prepared by the PD method was nearly comparable to that of LaMnO3/CeO2-MM, whereas the LaMnO3/CeO2-DI catalysts have a much smaller surface area than those prepared by the PD method. This implies that the DI of perovskite oxides in the pores of the CeO2 support promotes pore blockage during the calcination process.
We studied the LaMnO3/CeO2 catalysts using EXAFS to investigate the structure of the mixed oxides present on the CeO2 support. Figure 2 shows the La K-edge EXAFS spectra of LaMnO3/CeO2 catalysts along with those of the bulk LaMnO3 and La single oxide (La2O3). The EXAFS spectrum of the bulk LaMnO3 shows peaks at around 1.90 and 2.82 Ǻ, which were identified as the La–O and La–Mn contributions of the perovskite structure, respectively. The perovskite oxides have 12-coordinated La–O bonds with different bond lengths in their first coordination shells (2.50–3.01 Ǻ) and 8-coordinated La–Mn bonds in their second coordination shell (~3.37 Ǻ). The first coordination shell cannot be fitted by a single La–O bond, indicating that the shell was composed of several La–O bonds of different bond lengths. The peak in the second coordination shell, however, can be fitted by a single La–Mn bond. The spectrum of La–single oxide indicates a La–O contribution at 2.09 Ǻ and La–La contribution at 3.90 Ǻ, which were much different from the peak positions of the bulk LaMnO3.

La K-edge EXAFS spectra of LaMnO3/CeO2 catalysts and LaMnO3
The formation of a perovskite oxide phase for the LaMnO3/CeO2 catalysts was confirmed by the EXAFS studies. The LaMnO3/CeO2 catalysts also showed peaks at the same positions as those of the bulk LaMnO3 catalyst. The absence of peaks due to the La–O and La–La contributions in the spectra of the LaMnO3/CeO2 catalyst shows that there were no impurity phases in the supported catalysts. The curve fitting results for the EXAFS spectra of the LaMnO3/CeO2 catalysts are listed in Table 2. The peak for the La–Mn contribution at 282 Ǻ can be fitted well within the parameters of a single bond. The bond lengths for the La–Mn contribution were 3.340 Ǻ and were not influenced by the deposition method. The values were consistent with those for the bulk LaMnO3 perovskite oxides. The CN for the La–Mn contribution in LaMnO3/CeO2-PD was close to that for bulk LaMnO3, whereas the CN value was much lower for LaMnO3/CeO2-DI. Thus, the CN value for the perovskite oxide phase depends on the preparation method. Since the CN value is related to the crystalline sizes of LaMnO3, this implies that the sizes of the crystalline LaMnO3 of the LaMnO3/CeO2 catalyst were smaller than those of the bulk LaMnO3 catalyst and LaMnO3/CeO2-PD.
Figure 3 shows the XPS spectra of the LaMnO3/CeO2 catalysts and bulk LaMnO3. In the XPS spectra ranges of Mn 2p (635–660 eV), we observed the spin–orbit splitting of Mn 2p1/2 and Mn 2p3/2 signals (Fig. 4a). For LaMnO3, the peak position of the Mn 2p3/2 signal was located at around 641.1 eV (Table 1) [28]. In the case of LaMnO3/CeO2 prepared by the PD method, we observed peaks for the Mn 2p signals at the same positions as those for the bulk LaMnO3 catalyst. For LaMnO3/CeO2-DI, on the other hand, the Mn 2p3/2 peak was located in the range of 642.1 ± 0.1 eV, which was higher by 1.0 eV than that of bulk LaMnO3. The full width at half maximum (FWHM) for the Mn 2p3/2 signal was comparable and larger than that for bulk LaMnO3. This finding indicates that the oxidation state of Mn increased by the deposition of LaMnO3 on CeO2 by the DI method due to the strong interaction between LaMnO3 and CeO2.

XPS Mn 2p (a) and Ce 3d (b) spectra of LaMnO3/CeO2 catalysts and LaMnO3

TEM images of LaMnO3/CeO2-DI. Bright-field image (a), DES mapping image of La (b), Mn (c), and Ce (d)
In the Ce 3d region, peaks due to the presence of Ce 3d5/2 and Ce 3d3/2 were observed at 880–920 eV (Fig. 3b). These peaks mainly consisted of 3d5/2–3d3/2 spin–orbit-split doublets characteristic of stoichiometric CeO2 (Ce4+) [29]. The peak positions for the LaMnO3/CeO2 catalysts were the same as those for bulk LaMnO3, indicating that the surface states of the exposed Ce species were unaffected by the deposition of LaMnO3.
Table 1 lists the calculated Mn/Ce intensity ratios for the LaMnO3/CeO2 catalysts. The value was higher for the catalyst prepared by the DI method than that prepared by the PD method. Because the XPS peak intensity ratio of metal-support elements is related to the dispersion of the metal oxides on the support [30], we can conclude that the dispersion of the LaMnO3 phase was higher for LaMnO3/CeO2-DI. On the other hand, the XPS intensity ratio for LaMnO3/CeO2-PD was comparable to that for the mechanically mixed LaMnO3-CeO2.
Figure 4 shows TEM images of LaMnO3/CeO2-DI. The bright-field image (Fig. 2a) indicated that the catalyst was composed of particles with sizes of 10–20 nm. The DES mapping images (Fig. 2b–d) revealed that La and Mn were distributed over the whole particles of CeO2 support. These observations confirm that LaMnO3 particles were dispersed on the CeO2 for LaMnO3/CeO2-DI.
3.2 H2-TPR Studies
TPR of the LaMnO3/CeO2 catalysts by H2 was carried out to further investigate the structure and reducibility of the catalysts. Figure 5 shows the H2-TPR profiles for the LaMnO3/CeO2 catalysts, bulk LaMnO3, and CeO2. Here, the H2 consumption was normalized to 0.2 g and 0.8 g for LaMnO3 and CeO2, respectively, whereas the value was normalized to 1.0 g for LaMnO3/CeO2 catalysts. For the bulk LaMnO3 catalyst, H2 consumption was observed in temperature ranges of 200–400 °C and 700–900 °C. The peak in the lower temperature range was ascribed to the reduction of Mn4+ to Mn3+, and that in the higher temperature range to the reduction of Mn3+ to Mn2+ [10]. For the bulk CeO2 support, reduction peaks were observed at 300–550 °C due to the reduction of surface Ce4+ and at higher temperatures due to the reduction of the bulk CeO2 support [31]. In the case of LaMnO3/CeO2-PD, peaks appeared at around 136 °C. No peaks for the reduction of surface oxygen species on CeO2 were observed, which had been observed at around 500 °C for bulk CeO2. Quantitative analysis revealed that the amounts of H2 consumed in the range of 50–550 °C in the LaMnO3/CeO2 catalyst were larger than the sum of the values consumed for the bulk catalysts (0.20 g-LaMnO3 and 0.80 g-CeO2). This reveals that the peak for the surface oxygen species on CeO2 was shifted to a lower temperature by the LaMnO3 deposition and indicates that the reactivity of the surface oxygen species on CeO2 was improved. The peak maximum at around 800 °C was also slightly shifted to a lower temperature, indicating that the reactivity of the oxygen species on the catalysts and in the lattice were improved by the deposition of LaMnO3 onto CeO2.

H2-TPR profiles of LaMnO3/CeO2 catalysts and LaMnO3. Sample weight 0.050 g, gas composition 5 %H2–N2 balance, gas flow rate 30 mL/min, heating rate 10 °C/min
In the case of LaMnO3/CeO2-DI, the peaks in the low temperature range (~400 °C) were much different from those of the bulk LaMnO3 and LaMnO3/CeO2-PD: the H2 consumption started in the same temperature range as that of LaMnO3/CeO2-PD, although the peak at 136 °C increased and the temperature of the reduction peak for Mn4+ to Mn3+ for LaMnO3 was greatly lowered by the LaMnO3 deposition on CeO2. These findings indicate that, due to the strong interaction between LaMnO3 and CeO2, the reactivity of the oxygen species on the catalyst surface and in the lattice were improved by the deposition of LaMnO3 onto CeO2. The amount of H2 consumed in the range of 50–550 °C was larger than that consumed with LaMnO3/CeO2-PD. These results also indicate that the amount of H2 consumed is correlated with the dispersion of the perovskite oxides and that the interaction between LaMnO3 and CeO2 was controlled by changing the deposition method.
3.3 Catalytic Activity of CeO2-Supported Perovskite Oxides
Figure 6 shows the catalytic activities of supported LaMnO3 catalysts for NO oxidation in the temperature range of 200–500 °C, as compared with those for the bulk LaMnO3 catalyst. For the purposes of comparison, the conversion curves for the equilibrium of NO oxidation (NO + 1/2O2 ⇌ NO2) were indicated. The LaMnO3/CeO2 catalyst exhibited steady-state activity and NO was quantitatively transformed to NO2. The NO conversion increased with increased catalyst temperature up to 300 °C and decreased in the higher temperature range, following the equilibrium conversions. This indicates that the oxidation of NO to NO2 is equilibrated with the backward reaction. Thus, the activity for NO oxidation was dominated by not only kinetics but also thermodynamics. The bulk LaMnO3 catalyst exhibited lower NO conversion activity, with a maximum at 350 °C. The conversions of the CeO2 catalyst were lower than those for the LaMnO3 catalyst, with a maximum of 40 % conversions at 400 °C. A simple calculation shows that the activities of the LaMnO3/CeO2 catalyst cannot be explained in terms of the activities of bulk LaMnO3 and CeO2: the physical mixing of LaMnO3 and CeO2 would correspond to much lower conversions. The LaMnO3/CeO2-DI exhibited higher activity than the catalyst prepared by the PD method, even though the latter had a larger surface area. The rates for NO oxidation at 200 °C for LaMnO3/CeO2-DI and LaMnO3/CeO2-PD were 7.4 × 10−6 and 3.3 × 10−6 mol g−1 min−1, respectively. Thus, the high catalytic activity of LaMnO3/CeO2 prepared by the DI method was ascribed to the highly-dispersed LaMnO3 species on CeO2 and to the interaction between the catalytic materials. This result is probably due to the improved reactivity of the oxygen species of LaMnO3 by deposition on CeO2.

Catalytic activities of LaMnO3/CeO2 catalysts and LaMnO3 for oxidation of NO to NO2. Dashed line refers to the dependence of equilibrium conversion reaction temperature. Reaction gas NO 500 ppm-O2 5 %-N2 balance, catalyst weight 0.10 g, the gas flow 150 mL/min (W/F = 0.04 g s/mL)
Because the LaMnO3/γ-Al2O3 catalyst has shown high catalytic activity in propane oxidation [25], the results described above prompted us to compare the catalytic properties of the LaMnO3/CeO2 catalyst with those of a LaMnO3/γ-Al2O3 catalyst prepared by the incipient wetness method, in which LaMnO3 is deposited inside the alumina pores. The LaMnO3/CeO2 catalyst showed higher activity than the LaMnO3/γ-Al2O3 catalyst (Fig. 6), even though the surface area of the LaMnO3/CeO2 catalyst was much smaller than that of the LaMnO3/γ-Al2O3 catalyst (Table 1).
Lastly, we investigated the thermal stability of the LaMnO3/CeO2 catalyst for NO oxidation. The catalysts were heated at 850–1050 °C for 5 h in air and were again used for NO oxidation. As shown in Fig. 7, the X-ray diffraction (XRD) patterns of the LaMnO3/CeO2 catalyst exhibited the same CeO2 structure as calcined catalysts when the catalyst was calcined at 850 °C. As the calcination temperature increased, the peak intensities increased and the FWHM decreased, which indicates the sintering of the supports. Correspondingly, the catalyst surface area significantly decreased: the surface areas of the LaMnO3/CeO2 catalysts after calcination at 850, 950, 1050 °C were 24, 11, 2 m2/g, respectively. On the other hand, while the peaks for the perovskite oxides became more prominent, no other impurity phases were observed. It is worth noting that the calcination temperature in the range of 650–1050 °C was suitable for preparing bulk LaMnO3 perovskite powder, and the supported LaMnO3 phase was formed in the same temperature range.

XRD patterns of LaMnO3/CeO2-DI calcined at different temperatures
The NO oxidation activity in the thermally treated LaMnO3/CeO2 catalyst decreased with increased catalyst calcination temperature. The maximum temperature for NO oxidation shifted to a higher temperature and the maximum conversion decreased: the conversions slightly decreased after the calcination temperatures reached 850–950 °C, and significantly decreased after the calcination temperature reached 1050 °C (Fig. 8). We also investigated the themal stability of LaMnO3/γ-Al2O3 catalysts because they showed high stability for propane oxidation even when heated at high temperatures (650–1050 °C). Although the NO oxidation activity of the LaMnO3/γ-Al2O3 catalyst decreased after themal treatment at temperatures between 850–1050 °C, the decrease in the NO oxidation activity of these catalyst was smaller than in the LaMnO3/CeO2 catalysts. This is probably due to the improved stability of the perovskite phases when they were deposited inside the γ-Al2O3 pores. A comparison of the activity of the LaMnO3/CeO2 catalyst with that of LaMnO3/γ-Al2O3, showed that the LaMnO3/CeO2 catalyst exhibited higher activity after calcination at 850 °C, whereas this activity dropped significantly after calcination at 1050 °C. This decrease in the catalyst activity was probably due to the decreased surface area and catalyst sintering. Thus, the LaMnO3/CeO2 catalyst is effective in promoting NO oxidation when calcined in the temperature range of 650–850 °C.

Effect of heating treatment of LaMnO3/CeO2 and LaMnO3/γ-Al2O3, catalysts on their NO oxidation activities. The reaction condition was the same as that in Fig. 6
4 Conclusion
In this study, we prepared LaMnO3/CeO2 catalysts with different perovskite dispersions using four methods. The formation of LaMnO3 perovskite phases was confirmed by EXAFS studies. The dispersion of LaMnO3 perovskite oxide was evaluated by XPS. The LaMnO3/CeO2 catalyst prepared by the incipient wetness method exhibited higher NO oxidation activity although this catalyst has the smallest surface area. Based on the H2-TPR study results, we propose that the higher level of activity may be ascribed to the strong interaction between the perovskite phase and the CeO2 support. The supported catalyst was calcined in the temperature range of 650–1050 °C and then used for NO oxidation because thermal stability of the catalyst is important from a practical application perspective. The LaMnO3/CeO2 catalyst dry-impregnation method was more stable than LaMnO3/γ-Al2O3 catalyst against heat treatment at the temperature lower than 950 °C.
References
Olsson L, Persson H, Fridell E, Skoglundh M, Andersson B (2001) J Phys Chem B 105:6895
Després J, Elsener M, Koebel M, Kröcher O (2004) Appl Catal B 50:73
Bhatia D, McCabe R W, Harnold M P, Balakotaiah V (2009) J Catal 266:106
Peña MA, Fierro JLG (2001) Chem Rev 101:1981
Zhu J, Li H, Zhong L, Xiao P, Xu X, Yang X, Zhao Z, Li J (2014) ACS Catal 4:2917
Royer S, Duprez D, Can F, Courtois X, Batiot-Dupeyrat C, Laassiri S, Alamdari (2014) Chem Rev 114:10292
Kim CH, Qi G, Dahlberg K, Li W (2010) Science 327:1624
Chen J, Shen M, Wang X, Wang J, Su Y, Zhao Z (2013) Catal Commun 37:105
Qi G, Li W (2012) Catal Today 184:72–77
Chen J, Shen M, Wang X, Qi G, Wang J, Li W (2013) Appl Catal B Environ 134–135:251
He X, Meng M, He J, Zou Z, Li X, Li Z, Jiang Z (2010) Catal Commun 12:165
Zhou C, Liu X, Wu C, Wen Y, Xue Y, Chen R, Zhang Z, Shan B, Yin H, Wang WG (2014) Phys Chem Chem Phys 16:5106
Choi SO, Penninger M, Kim CH, Schneider WF, Thompson LT (2013) ACS Catal 3:2719
Dong Y-H, Xian H, Lv J-L, Liu C, Guo L, Meng M, Tan Y-S, Tsubaki N, Li X-G (2014) Mater Chem Phys 143:578
Wen Y, Zhang C, He H, Yu Y, Teraoka Y (2007) Catal Today 126:400
Wang J, Su Y, Wang X, Chen J, Zhao Z, Shen M (2012) Catal Commun 25:106
Zhong S, Sun Y, Xin H, Yang C, Chen L, Li X (2015) Chem Eng J 275:351
Cimino S, Colonna S, De Rossi S, Faticanti M, Lisi L, Pettiti I, Porta P (2002) J Catal 205:309
Mizuno N, Fujii, Igarashi H, Misono M (1992) J Am Chem Soc 114:7151
Chagas CA, Toniolo FS, Magalhães RNSH, Schmal M (2012) Int J Hydrog Energy 37:5022
You R, Zhang Y, Liu D, Meng M, Jiang Z, Zhang S, Huang Y (2015) Chem Eng J 260:357
Nguyen SV, Szabo V, Trong On D, Kaliaguine S (2002) Micropor Mesopor Mater 54:51
Xiao P, Hong J, Wang T, Xu X, Yuan Y, Li J, Zhu J (2013) Catal Lett 143:887
Asada T, Kayama T, Kusaba H, Einaga H, Teraoka Y (2008) Catal Today 139:37
Asada T, Kusaba H, Einaga H, Teraoka Y (2015) Bull Chem Soc Jpn 88:1036
Teraoka Y, Kakebayashi H, Moriguchi I, Kagawa S (1991) Chem Lett 20:673
Ankudinov AL, Ravel B, Rehr JJ, Conradson SD (1998) Phys Rev 58:7565
Zampieri G, Abbate M, Prado F, Caneiro A, Morikawa E (2002) Physica B 320:51
Holgado JP, Munuera G, Espinós JP, González-Elipe AR (2000) Appl Surf Sci 158:164
Briggs D (1976) J Electron Spectrosc Relat Phenom 9:487
Bueno-López A (2014) Appl Catal B 146:1
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Einaga, H., Yoshida, W., Lee, C. et al. Catalytic Properties of CeO2-Supported LaMnO3 for NO Oxidation. Catal Lett 146, 2495–2503 (2016). https://doi.org/10.1007/s10562-016-1878-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-016-1878-3