
Abstract
In this investigation, Mn-doped CeO2(CM) catalysts were prepared by co-precipitation (CP), sol-gel (SG), and hydrothermal (HT) methods and characterized by XRD, ICP-OES, N2 adsorption–desorption, SEM, TEM-HREM, Raman, XPS, FT-IR, XPS and H2-TPR techniques, and evaluated for CO oxidation activity. The CM catalyst obtained by HT method showed superior CO oxidation performance at much lower temperature (T50 = ~355 K). The better performance of CM-HT catalyst is ascribed to its larger surface area, higher concentration of surface adsorbed oxygen, enhanced reducibility, and more oxygen vacancy sites. Further, it was found that the CM-HT catalyst is fully recyclable for over four runs while maintaining its high activity, which suggests that it is a better catalyst for CO oxidation at lower temperatures.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Carbon monoxide (CO) is one of the important constituents of air pollutants from industrial plants and motor vehicles and it has been always major concern for environment as well as human health [1]. It was moderately stable with an atmospheric lifetime of 2–4 months and also a poisonous inhalant that deprives human body tissues of necessary oxygen [2, 3]. It also affects vegetation by interfering with plant respiration and nitrogen fixation [4]. The associated problems have driven the global scientific community to find out an effective method for the removal of CO. Though catalytic oxidation has been envisioned as one of the most appropriate methods for CO removal, development of the most effective oxidation catalyst is still under intense investigation [5]. This “simple” CO oxidation reaction is also highly valuable from the stand point of understanding many fundamental concepts in heterogeneous catalysis. Noble metal-based catalysts have been used typically for CO oxidation in automotive industry because of their excellent catalytic activity and desirable thermal stability [6, 7]. However, high cost and susceptibly to poisoning are major issues in this filed. Thus, it is very important to find out an alternative catalyst for the CO oxidation [8].
Over the past few decades, the rare-earth metal oxides have been widely used in environmental catalysis. Cerium oxide (CeO2) is particularly interesting because of its higher oxygen storage capacities (OSC) originated from easy switch over between cerium +3 and +4 oxidation states, thereby formation of labile oxygen vacancies and high mobility of oxygen from bulk to surface [9]. However, the most unavoidable problem associated with pure CeO2 is its poor thermal stability, high light off temperature, and less textural stability [10]. The catalytic efficiency of pure CeO2 reduces at elevated temperatures because of sintering and decline in surface area. Therefore, several studies have been focused on doped ceria catalysts, which showed significant enhancement in thermal stability, better redox properties, and higher catalytic activity [11, 12]. Recent reports suggest that the activity of pure CeO2 for complete oxidation reactions could be enhanced by the incorporation of cheaper transition metal oxides [13, 14]. Among the transition metal oxides, manganese oxide (MnOx) has been successfully employed as a doping agent for CeO2-based catalysts for various oxidation reactions including carbon monoxide [15], formaldehyde [15], toluene [16], benzene [17], soot particulates [18], hexane [19], and ethanol [20], as well as in the wet oxidation of aniline [21], phenol [22], acrylic acid [23], and volatile organic compounds [24]. In our previous study, we observed that CeO2-MnOx (CM) catalyst exhibits an excellent low temperature CO oxidation activity [25]. The improved oxidation activity is primarily attributed to an increase of ceria oxygen vacancy population, which may help to promote the migration of oxygen ions and the low-temperature redox ability through the formation of Ce–O–Mn solid solution [25]. Additionally, the structure and physicochemical properties of materials can be strongly dependent on the preparation method. Few investigations regarding the influence of different synthetic route on catalytic activity of Ce–Mn mixed oxide have been accounted in the literature. Arena et al. have demonstrated that, “redox-precipitation” route induce larger surface area and pore volume in nanostructured MnCeOx catalyst compared to simple co-precipitation, resulting in better activity and stability for CO oxidation [26]. Liu et al. has reported higher de-NOx activity of Mn–Ce catalyst prepared by surface template method than that of co-precipitation method, which has been attributed to higher surface area and smaller active particles leading to greater activation of NH3 and NOx molecules [27]. On the other hand, Shi et al. has accounted higher activity of Mn doped CeO2 catalyst prepared by modified co-precipitation method over co-precipitation and plasma method for CH4 conversion [28]. In the same line, Tang et al. has suggested that modified co-precipitation method included more surface Mn4+ species and richer lattice oxygen in the Mn–Ce catalyst than those prepared by sol–gel and co-precipitation methods, which in turn resulted in much higher catalytic activity towards complete oxidation of formaldehyde [29].
Taking into account the above considerations, the present work was specially focused on the specific structural features induced by different preparation methods, which facilitate the redox catalytic activity of ceria-manganese mixed oxides. With this aim, nanosized CM catalyst has been prepared by adopting three different methods, namely, (i) co-precipitation, (ii) sol–gel, and (iii) hydrothermal. The Ce/Mn mole ratio was fixed at 7/3. The obtained samples were subjected to thermal treatment at 773 K and were fully characterized by means of X-ray diffraction (XRD), inductively coupled plasma optical emission spectroscopy (ICP-OES), Brunauer–Emmett–Teller (BET) surface area, scanning electron microscopy (SEM), transmission electron microscopy (TEM), high resolution transmission electron microscopy (HRTEM), Raman spectroscopy (RS), X-ray photoelectron spectroscopy (XPS), Fourier-transform infrared (FT-IR) spectroscopy, and temperature programmed reduction by hydrogen (H2-TPR). Redox catalytic activity of the prepared catalysts has been investigated via oxidation of CO in the presence of oxygen. The relationship between structural features and CO oxidation activity, as well as the interaction between cerium and manganese ions has also been discussed.
2 Experimental Section
2.1 Catalysts Preparation
Nanosized CM catalyst, with the composition Ce0.7Mn0.3O2-δ (70:30 based on oxide ratio), were prepared by means of three different preparation routes. Stoichiometric amounts of Ce(NO3)3·6H2O and Mn(NO3)2·4H2O (supplied by Aldrich) as precursors were used in the synthesis.
2.2 Co-precipitation Method
An aqueous ammonia solution was added drop-wise to the Ce and Mn nitrate precursor solution until pH value reaches 9. Afterwards, the resulting suspension was left for 24 h and followed by the precipitate was filtered and thoroughly washed 4–5 times with de-ionized water to eliminate anion impurities. The obtained sample was then oven dried at 383 K for overnight and was ground with ceramic mortar. Finally, the well-dispersed powder was calcined for 5 h in a closed electric furnace at 773 K. The catalyst thus obtained was designed as CM-CP.
2.3 Sol–Gel Method
In this process, the urea was added to the Ce and Mn nitrate precursor solution as the complexation agent and the resultant mixed solution kept at 353 K for a few hours to ensure metal-complex formation. In a following step, ethylene glycol was added to the reaction solution and the temperature was raised to 363 K to facilitate the polyesterfication step. The resulting gel was dried at 383 K overnight. Then the gel was submitted to decomposition at 573 K for 4 h. Finally, the powder was collected and ground in an agate mortar followed by calcination at 773 K. The catalyst was designed as CM-SG.
2.4 Hydrothermal Method
In this method, an aqueous ammonia solution was slowly added to the Ce and Mn nitrate precursor solution at room temperature until the pH value of the mixture reached 9 with continuous stirring. Subsequently, the as-obtained suspension was transferred to 20 mL Teflon-lined stainless steel autoclaves, sealed and thermally treated at 473 K for 24 h, and then the autoclaves were cooled naturally to room temperature. The products were harvested by pressure filtration, washed fully with distilled water, and dried at 353 K for 12 h. Finally, the dried sample was calcined at 773 K for 5 h, and denoted as CM-HT. For comparison, pure CeO2 was prepared by precipitation of Ce(NO3)3·6H2O with an aqueous ammonia solution. The obtained solid was then dried at 383 K for 12 h, and calcined at 773 K for 5 h. Pure MnOx was also prepared in a similar way by using Mn(NO3)2·4H2O as precursor.
2.5 Catalysts Characterization
X-ray powder diffraction patterns were acquired on an X’pert Pro MPD powder diffractometer (PANAlytical Company) equipped with a nickel-filtered Cu Kα (0.15418 nm) radiation source and a scintillation counter detector. The scattered intensity data were collected from 2θ values of 2–80° by scanning at 0.01° steps with a counting time of 1 s at each step. Crystalline phases were identified by matching with the International Centre for Diffraction Data Powder Diffraction File. The average crystallite size (D) of the samples was determined with the help of Scherrer equation from line broadening and the lattice parameters (a) were estimated through the formulae a = (h2 + k2 + l2)1/2 × (λ/2sinθ) using the intensity of the most prominent peak (111).
The chemical analysis of the prepared samples was performed by inductively coupled plasma optical emission spectroscopy (ICP-OES, Thermo Jarrel Ash model IRIS Intrepid II XDL, USA) to confirm the respective concentrations of elements in the system. For ICP analysis, approximately 50 mg of the sample was dissolved in a solution of 25 mL aqua regia and 475 mL distilled water. Then 10 mL of the above solution was diluted to 250 mL.
The surface area and pore size distribution of the as-prepared samples were determined by N2 adsorption–desorption isotherms at liquid N2 temperature (77 K) on a Micromeritics (ASAP 2000) analyzer. Specific surface area and pore size distribution were calculated by BET and Barrett–Joyner–Halenda (BJH) methods, respectively. Prior to the measurements, the samples were pre-treated in a vacuum oven at 473 K for 2 h to remove any residual moisture.
SEM studies were performed by using a Zeiss EVO® series electron microscope to examine surface morphology, and the energy dispersive X-ray analysis was used to determine the elemental composition of samples. The SEM was equipped with a LaB6 source and an EDAX/DX4 detector. The acceleration potential voltage was maintained at 15 kV and samples were metallised with gold.
The TEM-HREM studies were made on a TECNAIG2 TEM microscope equipped with a slow-scan CCD camera and at an accelerating voltage of 200 kV. Samples for TEM analysis were prepared by crushing the materials in an agate mortar and dispersing ultrasonically in ethyl alcohol. After well dispersion, a droplet was deposited on a copper grid supporting a perforated carbon film and allowed to dry. The specimen was examined under vacuum at room temperature.
Raman spectra were obtained at room temperature on a Horiba Jobin–Yvon HR 800 Raman spectrometer fitted with a confocal microscope and liquid-nitrogen cooled charge-coupled device (CCD) detector. The line at 632 nm of Ar+ ion (Spectra Physics) laser was used as an excitation source for the visible RS. The laser was focused on the sample under a microscope with the diameter of the analyzed spot being ∼1 μm. The acquisition time was adjusted according to the intensity of Raman scattering. The wavenumber values reported from the spectra are accurate to within 2 cm−1.
XPS measurements were performed on a K-α spectrometer (Thermo scientific) using a monochromatic Al-Kα (1486.7 eV) radiation source. The analysis was done at room temperature and the pressures were typically in the order of less than 10−8 Pa. The charging of samples was corrected by setting the binding energy of the adventitious carbon (C 1s) at 284.5 eV. The samples were out gassed overnight in a vacuum oven at 283 K before XPS measurements. All the binding energies were measured within a precision of ± 0.3 eV.
The FT-IR spectra were recorded on a Nicolet Nexus spectrometer over the range of 400–4000 cm−1. The samples were diluted with KBr at a ratio of about 1:20 before tests, and the mixtures were then ground to a fine powder using an agate mortar and pestle. After finely mixed, the diluted samples were compressed into tablet form and then put on a holder for FT-IR spectral measurement.
Temperature programmed reduction analysis was used for evaluating the reduction properties of prepared catalysts using an automated Micromeritics AutoChem II-2720 instrument equipped with a thermal conductivity detector. Approximately 30 mg of the sample mass was placed on top of quartz wool in a U-shaped quartz reactor (Inner diameter 5 mm). The sample was pre-treated at a rate of 10 K min−1 to 473 K under a pure He atmosphere at a flow rate of 30 mL min−1 for 30 min to clean the surface of the catalysts before TPR investigation. After cooling down to room temperature, introducing the reduction agent of 5 vol % H2/Ar with a flow rate of 20 mL min−1, the temperature was programmatically raised to 1073 K at a ramp of 5 K min−1, keeping all the parameters unchanged. The outlet of the reactor was connected to a cold trap to absorb the water molecules resulted by the reduction of the catalysts.
2.6 Catalytic Activity Studies
The potential oxygen storage capacity was measured using the oxygen release characteristics of the sample with the help of conventional thermogravimetry (TG) method by employing a commercial Netzsch TG–DTA (Luxx, STA, 409 PC, Germany) analyzer under repeated thermal treatments in the temperature window of 573–1073 K. The weight change of the sample during the thermal treatments was continuously monitored with TG in flowing nitrogen or synthetic air. Approximately 15 mg of ceria-manganese powder was heated in the steps of 5 K min−1 up to 1073 K in nitrogen atmosphere (50 mL min−1) in order to release oxygen from the sample. After 10 min of stabilization, synthetic air was introduced resulting to an increase in sample weight due to the uptake of oxygen from air. The observed weight loss during the second stage of thermal treatment is considered as total OSC of the samples represented in µmol O2/g catalyst.
The activity of the catalysts for the oxidation of CO was measured in the temperature range of 250–750 K at normal atmospheric pressure using a fixed-bed micro reactor at a heating ramp of 5 K min−1. The samples (approximately 100 mg) were placed in the reactor (2 mm of inner diameter and length of 250 mm) and diluted with quartz particles with the same particle size, in order to keep a constant volume and better temperature control in the catalytic bed. The reaction temperature was monitored and controlled by using a thermo-couple inserted into the middle of catalyst bed. The following gases and gas mixtures were used (supplied by Air Liquid): argon (>99.999 % purity), 9.98 % CO (CO purity, >99.997 %), and 10.2 % O2 (oxygen purity, >99.995 %) with argon as balance. The CO/O2 reactant feed ratio was 1, and partial pressures of CO and O2 were in the range of 10 mbar. The total gas flow rates were guarded by using three mass flow controllers and were in the range of 50–60 N mL min−1 (milliliters normalized to 273.15 K and 1 atm.). The CO and CO2 gas concentrations were measured using an Uras 14 infrared analyzer module, and the O2 concentration was measured using a Magnos 16 analyzer (Hartmann & Braun). Prior to the oxidation of CO, the catalyst was heated to 773 K in a 10.2 % O2/Ar gas mixture, with a heating ramp of 10 K min−1, and kept at the final temperature for 1 h. The oxidized sample was then purged with Argon to avoid gas phase reaction and cooled to the desired starting temperature. For comparison, the catalytic activities of hydrothermally prepared pure CeO2 and MnOx were also measured.
3 Results and Discussion
3.1 Characterization Studies
Figure 1 shows the powder diffraction patterns of CM catalyst, which was prepared by co-precipitation, sol–gel, and hydrothermal methods, respectively. By comparison to a XRD pattern of a standard CeO2 sample (JCPDS 34-0394); the XRD lines of all of the CM samples were well indexed [30]. It was found that the as-synthesized CM samples possess a CeO2 cubic fluorite crystal structure. Further, the absence of MnO2, Mn2O3, and MnO XRD lines indicates either all the Mn ions have entered into the cubic ceria crystal forming solid solution or else the manganese oxides are present in well dispersed state, which is beyond the instrument’s detection limit [18, 29, 31]. Interestingly, the CM-HT sample showed the ceria diffraction lines at higher 2θ with broader nature and lower intensity than those of CM-CP and CM-SG (Fig. 1, right panel). It indicates that the hydrothermal synthesis provided smaller CeO2 crystallites. To clarify this observation, the crystallite sizes are calculated and summarized in Table 1. The increasing order of crystallite sizes was; CM-HT < CM-CP < CM-SG. Additionally, it can be clearly seen that the lattice parameters of ceria for all the three samples are smaller than pure cubic CeO2 (0.5411 nm), indicating obvious lattice contraction of CeO2. This lattice contraction could be due to the incorporation of smaller Mnx+ (Mn2+ = 0.083 nm; Mn3+ = 0.065 nm; Mn4+ = 0.053 nm) ions into the CeO2 cubic lattice resulting in the formation of solid solution in between corresponding metal oxides through the bridged oxygen [32]. It is worthy to notice that the lattice parameter of ceria for the CM-HT is much smaller than that of the other CM-CP and CM-SG samples, suggesting higher lattice contraction degree for the hydrothermal CM sample. Since the composition of mixed oxides is very essential for the catalytic oxidation reactions, the elemental analysis of each synthesized sample was of fundamental importance. Table 2 shows the amount of Ce and Mn in each sample obtained by ICP-OES. It is observed that the experimental atom percentage is quite closer to the nominal values. For simplicity, the nominal stoichiometric formula will be referred to in this work.

Powder X-ray diffraction patterns of Ce0.7Mn0.3O2-δ (CM-CP), Ce0.7Mn0.3O2-δ (CM-SG), and Ce0.7Mn0.3O2-δ (CM-HT) samples calcined at 773 K
The N2 adsorption–desorption isotherms and pore size distributions of the CM mixed oxides prepared with different preparation methods are shown in Fig. 2a and b, respectively. The effect of preparation method on the surface area, pore volume, and the pore diameter of CM are shown in Table 1. It was observed that very different textural and structural features can be achieved by varying the synthesis method for a single composition of CM mixed oxide. As shown in Fig. 2a, based on the classification of IUPAC, the N2 adsorption–desorption isotherms for CM-CP, CM-SG, and CM-HT are corresponding to irreversible type IV adsorption isotherms, which indicates the presence of mesopores that originate from the inner aggregation of nanoparticles. Meanwhile, the non-limiting adsorption at a high P/P 0 is characteristic of type H3, which implies the slit-like pore structures of samples [33]. Further, the pore size distributions of the catalysts were measured from the N2 desorption isotherm using the cylindrical pore model (BJH method). All the samples showed unimodal pore size distribution and the size of pores are in the range of mesoporous nature. However, the pore size distribution of the resulting samples showed considerable difference with respect to the preparation method. The average distribution of mesopores for CM-CP, CM-SG, and CM-HT are ~30.1, 24.8, and 17.2 nm, respectively. Moreover, CM-HT shows the narrowest mesopores distribution, suggesting that the hydrothermal process favors the formation of more orderly and narrower distribution of mesopores. It is well acknowledged that, catalysts with small orderly pores and well dispersed particles are favorable for the improvement of catalytic performance. Generally, a high surface area has a beneficial effect on the catalytic activity for catalysts. Among all the catalysts, CM-HT catalyst shows the highest specific surface area (69 m2 g−1) and pore volume (0.47 cm3 g−1) while compared with that of other catalysts. The high surface area of CM-HT catalyst may be due to the existence of strong interaction between cerium and manganese oxides.

a N2 adsorption–desorption isotherms of Ce0.7Mn0.3O2-δ (CM-CP), Ce0.7Mn0.3O2-δ (CM-SG), and Ce0.7Mn0.3O2-δ (CM-HT) samples calcined at 773 K. b Pore size distribution curves of respective samples
The surface morphology of the as prepared CM-CP, CM-SG and CM-HT samples was studied by SEM and the obtained images are shown in Fig. 3a–c, respectively. The SEM micrographs clearly shows the obtained powders are of nanosized particles with a uniform size distribution. It is evident that the relatively small agglomerated spherical particles are obtained in case of both CM-CP and CM-HT samples, whereas the CM-SG sample consisted of quite larger particles with regular spherical morphology. Thus, the results evidenced that the preparation method slightly affected the size of the CM nanocatalysts, as agreed with XRD and BET results. It is very difficult to find out exactly the particle size of the catalysts from the SEM analysis. Hence, TEM micrographs of the samples are collected and the images of these catalysts are shown in Fig. 4a, c, and e. It can be seen that in all catalysts the nanosized particles are uniformly distributed with the interconnected spherical morphology along the surface, and the way of preparation method has little influence on the morphology of the products. However, there is a change in the particle size of the nanoparticles on each sample. On the basis of extensive image analysis, the average particle size of CM-CP, CM-SG and CM-HT samples are in the range of ~7–8, 9–10 and 5–6 nm, respectively. To gain further insight into the microstructural characteristics of different CM catalysts, HRTEM analysis has been performed to distinguish the relative pattern of the species. As shown in the HRTEM images of the three samples in Fig. 4b, d and f, there is only one type of lattice fringes with spacing’s of about 0.309 nm ascribed to the (111) crystallographic planes of CeO2 [34, 35]. On the other hand, no crystal planes of MnOx can be seen in all the CM mixed oxide samples, indicating the total incorporation of Mn into CeO2 lattice and forming the Ce–O–Mn solid solution particles. This is consistent with the observed (111) XRD peak.

SEM (a, b, c) images of Ce0.7Mn0.3O2-δ (CM-CP), Ce0.7Mn0.3O2-δ (CM-SG), and Ce0.7Mn0.3O2-δ (CM-HT) samples calcined at 773 K

TEM (a, c, e) and HRTEM (b, d, f) images of Ce0.7Mn0.3O2-δ (CM-CP) Ce0.7Mn0.3O2-δ (CM-SG), and Ce0.7Mn0.3O2-δ (CM-HT) samples calcined at 773 K
RS is a useful technique to confer information regarding the presence of oxygen vacancies and existence of different crystalline phases in metal oxide materials. Raman spectra are dominated by metal–oxygen lattice vibrations and are sensitive to the crystalline symmetry, in contrast to XRD results, which are related to the cation sublattice. Thus, RS provides complementary information to the XRD results [36]. Figure 5 shows the Raman spectra of all the CM catalysts. Spectrum for pure CeO2 is also included, for comparative purpose. At first glance as seen in Fig. 5, pure ceria and all the CM samples exhibit a prominent peak at about 451–465 cm−1, which is corresponding to the triply degenerate F2g vibrational mode of the perfect CeO2-based cubic fluorite lattice (the only one allowed in first order) [37]. It is well-known that the Raman shift is very sensitive to lattice distortion by dopants. It can be clearly seen from the figure that a small red shift along with a certain decrease in intensity is observed in the fluorite F2g band for the Mn-containing catalysts. This phenomenon indicates that the interaction of Ce and Mn deform the fluorite structure and forms the oxygen vacancies in the lattice in order to maintain the charge imbalance upon doping of Mn ions [38]. We cannot, however, discard that the shift is partially due to some effects including phonon confinement, strain and defects in the very small CeO2 crystallites present in these samples [39]. The formation of oxygen vacancies confirmed by the Raman mode at about ~646–653 cm−1 in the Mn-doped ceria catalysts [40]. The peak shifting accompanying with broadening in the case of CM-HT (~14 cm−1) was slightly more than that of the CM-CP (~11 cm−1) and CM-SG (~7 cm−1) which shows a higher concentration of oxygen vacancies in CM-HT than CM-CP and CM-SG samples. The shift in the position of F2g band and absence of manganese oxide peaks in all CM catalysts evidenced the formation of homogeneous solid solution as indicated by the XRD and HRTEM results.

Raman spectra of pure CeO2, Ce0.7Mn0.3O2-δ (CM-CP), Ce0.7Mn0.3O2-δ (CM-SG), and Ce0.7Mn0.3O2-δ (CM-HT) samples calcined at 773 K
It is well established that catalyst surface elemental composition and chemical states are very important during the process of catalytic reaction because they can strongly affect the catalytic efficiency. The surface composition and chemical states can be determined from an XP spectrum according to their characteristic binding energies on catalyst surfaces. Hence, XPS core level spectra of Ce 3d, Mn 2p, and O 1s were· measured for investigated samples and the corresponding results are presented in Fig. 6a–c, respectively. These spectra have been calibrated against the C 1s peak standardized at 284.5 eV. Figure 6a shows the Ce 3d spectrum collected for CM mixed oxide samples. Generally, cerium compounds exhibit complex features due to strong hybridization with ligand orbitals and fractional occupancy of the valence 4f orbitals [41]. Based on the work of Yang et al. the deconvoluted Ce 3d spectrum can be assigned as follows [42]. Two sets of spin orbital multiplets, corresponding to the 3d3/2 and 3d5/2 contributions are labeled as u and v, respectively [42]. The six peaks labeled as v/u, v″/u″ and v′′′/u′′′ referring to three doublets of the spin–orbit split components are attributed to Ce4+ final states. The high binding energy doublet v′′′/u′′′ are attributed to the final state of Ce 3d9 4f0 O 2p6; doublet v″/u″ are attributed to the state of Ce 3d9 4f1 O 2p5, and doublet v/u correspond to the state of Ce 3d9 4f2 O 2p4. The other two peaks labeled as v′/u′ referring to one doublet of the spin–orbit split component is attributed to Ce3+ final state. The doublet v′/u′ are corresponding to the final state of Ce 3d9 4f1 O 2p6 [43, 44]. It can be seen that for all the Ce-containing samples, the cerium ions show a mixture of 3+ and 4+ oxidation states, indicating Ce3+ and Ce4+ ions co-exist at the surface of the catalysts. As the Ce3+ fraction is directly related to the concentration of oxygen vacancies generated, it can be assumed that the oxygen vacancy defect density is proportional to the Ce3+ fraction of each sample. The relative amount of Ce3+ in the ceria catalysts could be calculated by using an area ratio of the sum of the peaks derived from Ce3+ to the total area of the Ce 3d spectrum [45]. CM-HT exhibits higher relative concentration of [Ce3+]/[Ce3++Ce4+], compared with that of other catalysts, in good agreement with the results of Raman analysis. The relative concentration of [Ce3+]/[Ce3++Ce4+] is obtained in Table 3 in the sequence of CM-HT > CM-CP > CM-SG, indicating that there are more oxygen vacancies presenting in the ceria of CM-HT catalyst. In addition, the binding energy of peaks of CM-HT is lower than that of the other two catalysts; this might be caused by strong interactions between ceria and Mn particles thorough the formation of Ce–O-Mn solid solution. The core level spectra of Mn 2p (Fig. 6b) exhibited two peaks in the range of 640–658 eV and these peaks are corresponding to the Mn 2p3/2 and Mn 2p1/2 electron spin states (around 640 and 654 eV, respectively). However, the asymmetric nature of the Mn 2p peaks indicates the presence of Mn in different oxidation states. Hence, in order to find out the different valent Mn species, the Mn 2p3/2 peak could be satisfactorily deconvoluted (using Gaussian peak fitting) into three peaks at binding energies of 640.4–640.7, 641.6–642.1 and 643.4–644.5 eV, which corresponded to Mn2+, Mn3+ and Mn4+, respectively [46]. Thus, the Mn 2p XPS analysis conclusively establishes the presence of Mn4+ together with Mn2+ and Mn3+ ions in all the samples. It has been commonly observed that the high oxidation state of Mn species on the catalyst surface is favorable for the oxidation of CO over the manganese containing catalysts [47]. The amount of Mn4+/Mntotal species on the catalyst surface (relative abundance, at %) decreases as: CM-HT (45.71) > CM-CP (43.26) > CM-SG (40.58). Interestingly, the Mn 2p core level features in the CM-HT are shifted to lower binding energy compared with other samples. This shift in binding energy and the higher surface atomic ratio of Mn4+/Mntotal in CM-HT may be attributed to the presence of strong interaction between the Ce- and Mn-oxides or a well dispersion of manganese oxide particles.

a Ce 3d, b Mn 2p, c O 1s, d FT-IR spectra of Ce0.7Mn0.3O2-δ (CM-CP), Ce0.7Mn0.3O2-δ (CM-SG), and Ce0.7Mn0.3O2-δ (CM-HT) samples calcined at 773 K
O 1s XPS results of CM-CP, CM-SG and CM-HT are shown in Fig. 6c. The O 1s peaks showed an asymmetric shape and were deconvoluted into three sub peaks in each sample by Gaussian peak fitting. The first sub peak (labeled as Oa) appeared about at 529.2–529.7 eV could be assigned to the lattice oxygen in the CM mixed oxide crystallites (Ce–O–Mn) [48]. This component provides further support for the formation of solid solution between Ce–O and Mn–O. The second sub-peak (labeled as Ob) at 530.7–531.9 eV can be attributed to hydroxyl groups and/or weakly surface adsorbed oxygen. And the other sub-peak (labeled as Oc) at 532.3–533.7 eV belongs to the water and/or carbonate species as contaminants on the surface [49]. The analysis of the FT-IR also evidenced the presence of water and carbonate species in the studied catalysts, as shown in Fig. 6d. Usually, the surface adsorbed oxygen is considered to be more reactive than others due to their higher mobility, which can efficiently promote oxidation reactions at low temperatures [50]. To evaluate the proportion of Ob in these catalysts, the Ob/Ototal ratios were used and the obtained results are shown in Table 3. More importantly, one can see that the relative intensities of Ob were related closely to the preparation method of CM. As shown in Table 3, with respect to CM-SG (34.82 %) and CM-CP (36.11 %), CM-HT (39.47 %) possesses a higher ratio of surface adsorbed oxygen, which is reasonable because the surface adsorbed oxygen is closely bound to the Ce3+ ion.
To investigate the effects of Mn doping and preparation method on the intrinsic redox properties of the studied catalysts, the H2-TPR studies were carried out and the results are shown in Fig. 7a, b. It can be clearly seen from Fig. 7a that all CM catalysts yield three typical sequent reduction zones. Further, the H2-consumption peaks of all the samples could be attributed to the reduction of ceria or manganese species, and their reduction behavior is very different from each other. From Fig. 7b and in line with the results reported in the literature, pure ceria (hydrothermally prepared) displays two reduction peaks at around ∼702 and 1058 K, which are attributed to the surface reduction and the bulk reduction, respectively, following the idea that oxygen mobility is relatively slow within the bulk compared to surface of ceria [51]. Therefore, the low temperature peak (denoted as α) about at 498–514 K reveals the reduction of MnO2/Mn2O3 to Mn3O4 and the medium temperature peak (denoted as β) around 588–613 K assigned to reduction of Mn3O4 to MnO together with the oxygen removal from surface of mixed oxide [52]. Finally, the high temperature peak (denoted as γ) around at ~980–1050 K related to reduction of bulk oxygen, which suggests that the diffusivity of bulk oxygen in ceria significantly increases with the doping of Mn species [53]. Existence of synergistic interaction between Ce–O and Mn–O in the mixed oxides, which facilitates the movement of lattice oxygen from bulk to surface, elucidates the shift of reduction maxima to lower temperature. Further, the reduction behaviors of the CM catalysts are significantly influenced by the preparation method. It is worthy to stress that the CM-HT sample shows the lowest temperature for each reduction peak among the three samples, which is in accordance with its higher catalytic activity of CO oxidation (as shown in Fig. 8a). Additionally, the high specific surface area of CM-HT (Table 1) also should be beneficial to its reduction at low temperature. On the other hand, CM-CP and CM-SG also exhibit same reduction patterns, however the positions and intensities of the peaks are different from those of CM-HT, which may be due to the weak interactions between Ce–O and Mn–O oxides. Based on these results, it can be inferred that hydrothermal method bestows with higher concentration of well dispersed manganese oxide species in ceria, which facilitated the surface and bulk reduction of CeO2 at lower temperature in Ce-O-Mn solid solution.

H2 consumption as a function of temperature for pure CeO2 (HT), Ce0.7Mn0.3O2-δ (CM-CP), Ce0.7Mn0.3O2-δ (CM-SG), and Ce0.7Mn0.3O2-δ (CM-HT) samples calcined at 773 K

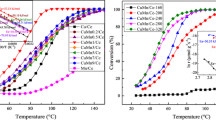
a Conversion of CO (%) versus temperature (K) for pure CeO2, MnOx, Ce0.7Mn0.3O2-δ (CM-CP), Ce0.7Mn0.3O2-δ (CM-SG), and Ce0.7Mn0.3O2-δ (CM-HT) samples calcined at 773 K. b CO conversion as a function of reaction temperature for four successive runs of Ce0.7Mn0.3O2-δ (CM-HT) sample calcined at 773 K
3.2 Catalytic Activity Studies
OSC is an important characteristic to determine the catalytic properties of a catalyst. It provides information about the most reactive and most available oxygen atoms in the catalyst. Table 3 shows the OSC values for the synthesized samples. It can be clearly seen that the OSC is affected by different preparation methods. The OSC of CM-HT is 235 (µmol O2/g), which is much higher than that of CM-CP (208 µmol O2/g) and CM-SG (175 μmol O2/g). The improved OSC in the hydrothermally prepared CM solid solution may be credited to delocalized oxygen vacancies, weakly bound oxygen, and oxygen intestines in their defective crystal structures. The incorporation of manganese (i.e. substitution of Ce4+ by the Mn ions) will lead to the formation of extrinsic oxygen vacancies (extrinsic defects) within the ceria lattice to maintain charge balance. Thus defects created during the incorporation of Mn ions lead to ease of formation of labile oxygen vacancies, connected to this relatively high mobility of bulk oxygen species within the lattice cell thereby enhancement in the OSC [54]. These results are consistent with the Raman and XPS results evidencing structure defects.
The low temperature catalytic oxidation of CO to CO2 has long been a benchmark reaction in heterogeneous catalysis due to its practical importance in many industrial processes [55]. The catalytic activity for CM catalysts, which was prepared by CP, SG, and HT methods together with the pure CeO2 and MnOx (comparison purpose), are graphically expressed by plotting conversion of CO as a function of reaction temperature in Fig. 8a. As observed, CO conversion increased with increasing reaction temperatures for CM-CP, CM-SG, and CM-HT catalysts. Further, it was found that the Mn-doped CeO2 samples displayed enhanced catalytic activity compared to pure CeO2 and MnOx. However, the activity of the doped catalysts with same composition was substantially influenced by the preparation method. For comparison purpose, the temperatures at which the CO conversion reached 50 and 90 % (T50 and T90) were evaluated and summarized in Table 3. Among the three CM mixed oxides prepared by different methods, the CM-HT exhibited the highest catalytic activity and the activity ranking is in the order of CM-HT > CM-CP > CM-SG > CeO2 > MnOx. The T50 and T90 values obtained for CM-HT were ~355 and 405 K, respectively. The activity tests suggested that the hydrothermal process imparted superior physicochemical property to CM catalyst for CO oxidation over co-precipitation and sol–gel methods.
The diversity of low temperature catalytic activity over the CM-HT catalyst can be significantly related to the following factors: The incorporation of lower valent manganese ions into ceria lattice resulted in decreased crystallite size (more in CM-HT) which increased the lattice strain. This increased lattice strain was beneficial for the activation of reactant molecules on the surface [56]. Further, with incorporation of Mn into CeO2, the smaller nanoparticles (TEM) are obtained, which promoted the nano-effect of the particles. Generally speaking, for the complete oxidation of CO, the catalyst with a higher surface area exhibits a better catalytic activity. Both the smaller crystallite sizes (XRD) and mesoporous structure (BET) made a contribution to the highest surface area of CM-HT than CM-CP and CM-SG catalysts. It is understandable that CM-HT with large BET specific surface area can provide a larger number of active sites and consequently led to higher conversions of CO. The better performance can also be explained from the surface oxygen vacancies. From XPS results it is clear that, the CM-HT achieved more number of oxygen vacancies (i.e., Ce3+ species), which provides freedom for the movement of lattice oxygen, thus increased the mobility of oxygen in the surface as well as bulk ceria and improved the catalytic performance [57]. This is further supported by Raman and H2-TPR results (Figs. 5, 7) that provide direct spectroscopic evidence that the high catalytic activity of the CM-HT sample is related to the high concentration of surface oxygen vacancy sites (whereby the Ce3+ ions are formed). From Table 3, it can be clearly seen that the fractions of the surface adsorbed oxygen component in the CM-HT catalyst is relatively high. There is also a great quantity of Mn4+ ions existing on the surface of CM-HT, as shown in Table 3. It is well recognized in the literature that the surface adsorbed species from defective sites with an unsaturated structure and the higher valence oxidation state of Mn species are of great importance in the catalytic CO oxidation process [58, 59]. It is generally accepted that OSC is dependent on specific surface area; however, the OSC/BET value of CM-HT sample is less (3.41) while compared to CM-CP (3.59) and CM-SG (3.57) values. In contrast, CM-HT sample showed highest catalytic activity. Therefore, we can conclude that catalytic activity solely not dependent on the specific surface area. This might be due to other factors such as high OSC and the presence of more oxygen vacancies. All these results from the structural characterizations and CO oxidation activity study demonstrated that the hydrothermal process bestowed superior CO oxidation activity on the CM catalyst over co-precipitation and sol–gel methods. The promising activity of the CM-HT catalyst encouraged us to further investigate its stability during repetitive runs. To evaluate the stability of CM-HT catalyst, CO oxidation for four successive runs were investigated and the results are presented in Fig. 8b. Even though the catalytic activity slightly decreased upon repeated CO oxidations, a significant deactivation was not observed in Fig. 8b. This finding verified the excellent stability and prominent catalytic activity of the as-prepared CM-HT catalyst.
4 Conclusions
In summary, three manganese doped-CeO2 catalysts were prepared by employing co-precipitation, sol–gel and hydrothermal methods, respectively. The catalytic activity of the oxides was investigated for CO oxidation reaction. The XRD results showed that the as synthesized CM catalysts have similar cubic fluorite structures and the composition values of the elements present in the catalysts were confirmed by ICP-OES. The nanocrystalline texture of mixed metal oxides is clearly evidenced by TEM investigation. Raman measurements suggest a defective structure of the mixed oxides resulting in the formation of oxygen vacancies. XPS analysis reveal that Ce is in the form of Ce4+ and Ce3+ (oxygen vacancies), and Mn existed in the form of Mn4+, Mn3+, and Mn2+ on the surface of the sample. The reducibility of ceria has been increased upon doping with Mn cations and this enhancement is more in case of sample prepared by the hydrothermal method. All the CM mixed oxide catalysts were highly active for CO oxidation at low temperature compared with that of pure ceria and manganese oxides. However, difference in preparation method was found to influence the activity. The sequential order of the catalytic activity is as follows: CM-HT > CM-CP > CM-SG. The superior catalytic performance of CM-HT (T50 = ~355 K) is mainly accredited to the stronger synergistic interaction between ceria and manganese components, the formation of Ce–O–Mn solid solution, the high surface area, and unique pore structure. Additionally, the existence of a large number of surface adsorbed oxygen species, Mn4+ species and oxygen vacancies (Ce3+) present on the surface promoted the activity. Moreover, the stability of the catalyst was found to be satisfactory. These results showed the potential characters of CM-HT nanoparticles are suitable to be applied in three-way catalysts.
References
Schneidemesser E, Monks PS, Plass-Duelmer C (2010) Atmos Environ 44:5053–5064
Choudhary TV, Sivadinarayana C, Chusuei CC, Datye AK, Fackler JP, Goodman DW (2002) J Catal 207:247–255
Imanaka N, Masui T, Imadzu H, Yasuda K (2011) Chem Commun 47:11032–11034
Prasad R, Singh P (2012) Catal Rev Sci Eng 54:224–279
Royer S, Duprez D (2011) Chem Cat Chem 3:24–65
Lakshmi K, Thrimurthulu G, Reddy BM, Muhler M, Grunert W (2011) Catal Sci Technol 1:1645–1652
Wang H, Zhu H, Qin Z, Liang F, Wang G, Wang J (2009) J Catal 264:154–162
Everaert K, Baeyens J (2004) J Hazard Mater 109:113–139
Shan W, Liu F, Yu Y, He H (2014) Chin J Catal 35:1251–1259
Lakshmi K, Sudarsanam P, Thrimurthulu G, Reddy BM (2010) Appl Catal B 101:101–108
Hilaire S, Luo L, Rechberger F, Krumeich F, Niederberger M (2014) Z Anorg Allg Chem 640:733–737
Sasikala R, Gupta NM, Kulshreshtha SK (2001) Catal Lett 71:69–73
Zhen J, Wang X, Liu D, Song S, Wang Z, Wang Y, Li J, Wang F, Zhang H (2014) Chem Eur J 20:4469–4473
Su Y, Wang S, Zhang T, Wang S, Zhu B, Cao J, Yuan Z, Zhang S, Huang W, Wu S (2008) Catal Lett 124:405–412
Xuesong L, Jiqing L, Kun Q, Weixin H, Mengfei L (2009) J Rare Earths 27:418–424
Liao Y, Fua M, Chena L, Wu J, Huang B, Ye D (2013) Catal Today 216:220–228
Wang Z, Shen G, Li J, Liu H, Wang Q, Chen Y (2013) Appl Catal B 138–139:253–259
Venkataswamy P, Jampaiah D, Rao KN, Reddy BM (2014) Appl Catal A 488:1–10
Picasso G, Gutierrez M, Pina MP, Herguido J (2007) Chem Eng J 126:119–130
Rao T, Shen M, Jia L, Hao J, Wang J (2007) Catal Commun 8:1743–1747
Levi R, Milman M, Landau MV, Brenner A, Herskowitz M (2008) Environ Sci Technol 42:5165–5170
Chen H, Sayari A, Adnot A, Larachi F (2001) Appl Catal B 32:195–204
Silva AMT, Marques RRN, Quinta-Ferreira RM (2004) Appl Catal B 47:269–279
Delimaris D, Ioannides T (2008) Appl Catal B 84:303–312
Venkataswamy P, Rao KN, Jampaiah D, Reddy BM (2015) Appl Catal B 162:122–132
Arena F, Trunfio G, Fazio B, Negro J, Spadaro L (2009) J Phys Chem C 113:2822–2829
Liu Z, Yi Y, Zhang S, Zhu T, Zhu J, Wang J (2013) Catal Today 216:76–81
Shi L, Chu W, Qu F, Luo S (2007) Catal Lett 113:59–64
Tang X, Li Y, Huang X, Xu Y, Zhu H, Wang J, Shen W (2006) Appl Catal B 62:265–273
Tang Y, Qiao H, Wang H, Tao P (2013) J Mater Chem A 1:12512–12518
Mingshan C, Yuan L, Xinquan W, Jun W, Meiqing S (2013) J Rare Earths 31:572–576
Tang X, Chen J, Huang X, Xu Y, Shen W (2008) Appl Catal B 81:115–121
Sing KSW, Everett DH, Haul RAW, Moscou L, Pierotti RA, Rouquerol J, Siemieniewska T (1985) Pure Appl Chem 57:603–619
Singh P, Hegde MS (2010) Dalton Trans 39:10768–10780
Yanan D, Jiansen N, Pengfei H, Junan W, Xueling H, Hui X (2013) J Rare Earths 31:271–275
Lan L, Chen S, Cao Y, Gong M, Chen Y (2015) Catal Sci Technol 5:4488–4500
Shyu JZ, Weber WH, Gandhi HS (1988) J Phys Chem 92:4964–4970
McBride JR, Hass KC, Poindexter BD, Weber WH (1994) J Appl Phys 76:2435–2441
Choudhury B, Choudhury A (2012) Mater Chem Phys 131:666–671
Barros FAA, Sousa HSA, Oliveira AC, Junior MC, Filho JM, Viana BC, Oliveira AC (2013) Catal Today 212:127–136
Romeo M, Bak K, Fallah JE, Normand FL, Hilaire L (1993) Surf Interface Anal 20:508–512
Yang L, Tan Y, Sheng Z, Zhou A (2014) J Nanomater 2014:368583–368588
Ozkara-Aydinoglu S, Ozensoy E, Aksoylu AE (2009) Int J Hydrogen Energy 43:9711–9722
Jampaiah D, Ippolito SJ, Sabri YM, Reddy BM, Bhargava SK (2015) Catal Sci Technol 5:2913–2924
Devaiah D, Tsuzuki T, Aniz CU, Reddy BM (2015) Catal Lett 145:1206–1216
Zhao P, Wang C, He F, Liu S (2014) RSC Adv 4:45665–45672
Zou Z-Q, Meng M, Zha Y-Q (2010) J Phys Chem C 114:468–477
Qi L, Yu Q, Dai Y, Tang C, Liu L, Zhang H, Gao F, Dong L, Chen Y (2012) Appl Catal B 119–120:308–320
Zhang D, Zhang L, Shi L, Fang C, Li H, Gao R, Huang Zhang L (2013) Nanoscale 5:1127–1136
Cai S, Zhang D, Zhang L, Huang L, Li H, Gao R, Shi L, Zhang J (2014) Catal Sci Technol 4:93–101
Zhang G-F, Li L-P, Li G-S, Qiu X-Q (2010) Chi J Struct Chem 29:307–313
Tang W, Wu X, Li D, Wang Z, Liu G, Liu H, Chen Y (2014) J Mater Chem A 2:2544–2554
Wu X, Liang Q, Weng D, Fan J, Ran R (2007) Catal Today 126:430–435
Chen H, Sayari A, Adnot A, Larachi F (2001) Appl Catal B 32:195–204
Freund H-J, Meijer G, Scheffler M, Schlögl R, Wolf M (2011) Angew Chem Int Ed 50:10064–10094
Yao X, Tang C, Ji Z, Dai Y, Cao Y, Gao F, Dong L, Chen Y (2013) Catal Sci Technol 3:688–698
Yuejuan W, Jingmeng M, Mengfei L, Ping F, Mai H (2007) J Rare Earths 25:58–62
Deng J, Zhang L, Dai H, He H, Au CT (2008) Catal Lett 123:294–300
Zou ZQ, Meng M, Zha YQ (2010) J Phys Chem C 114:468–477
Acknowledgments
We greatly acknowledge Prof. Dr. W. Grünert, Ruhr University Bochum, Germany for providing CO oxidation results. PV and DM thank Council of Scientific and Industrial Research (CSIR), and United Grant Commission (UGC), New Delhi for the Senior and Junior Research Fellowships respectively. DJ thanks IICT-RMIT Joint Research Centre for the award of Junior Research Fellowship. Financial support was received from the Department of Science and Technology, New Delhi, under SERB Scheme (SB/S1/PC-106/2012).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Venkataswamy, P., Jampaiah, D., Mukherjee, D. et al. Mn-doped Ceria Solid Solutions for CO Oxidation at Lower Temperatures. Catal Lett 146, 2105–2118 (2016). https://doi.org/10.1007/s10562-016-1811-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-016-1811-9