
Abstract
Introduction
Transplantation of a cell-seeded graft may improve wound healing after radiotherapy. However, the survival of the seeded cells depends on a rapid vascularization of the graft. Co-culturing of adult stem cells may be a promising strategy to accelerate the vessel formation inside the graft. Thus, we compared the in vivo angiogenic potency of mesenchymal stem cells (MSC) and endothelial progenitor cells (EPC) using dorsal skinfold chambers and intravital microscopy.
Materials and methods
Cells were isolated from rat bone marrow and adipose tissue and characterized by immunostaining and flow cytometry. Forty-eight rats received a dorsal skinfold chamber and were divided into 2 main groups, irradiated and non-irradiated. Each of these 2 groups were further subdivided into 4 groups: unseeded matrices, matrices + fibroblasts + pericytes, matrices + fibroblasts + pericytes + MSCs and matrices + fibroblasts + pericytes + EPCs. Vessel densities were quantified semi-automatically using FIJI.
Results
Fibroblasts + pericytes − seeded matrices showed a significantly higher vascular density in all groups with an exception of non-irradiated rats at day 12 compared to unseeded matrices. Co-seeding of MSCs increased vessel densities in both, irradiated and non-irradiated groups. Co-seeding with EPCs did not result in an increase of vascularization in none of the groups.
Discussion
We demonstrated that the pre-radiation treatment led to a significant decreased vascularization of the implanted grafts. The augmentation of the matrices with fibroblasts and pericytes in co-culture increased the vascularization compared to the non-seeded matrices. A further significant enhancement of vessel ingrowth into the matrices could be achieved by the co-seeding with MSCs in both, irradiated and non-irradiated groups.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Soft tissue defects caused by trauma or tumor resection provide a challenging problem to reconstructive surgery and tissue engineering. Tissue engineering techniques offer the development of bioactive tissue constructs that can regenerate soft tissue in both structure and function (Choi et al. 2010). Cell-seeded biodegradable natural materials like acellular dermis may be used for soft tissue augmentation (Krauss 1999). Nonetheless, the absence of a vascular network within such a graft may have a negative effect on the viability of the seeded autologous cells in the graft due to an inadequate supply with nutrients and gas exchange after implantation (Laschke et al. 2006; Phelps and Garcia 2010). Thus, a rapid vascularization of the biomaterial plays a pivotal role in the survival and proliferation of the seeded cells to gain a successful integration of the graft into the host tissue. In addition, a neoadjuvant irradiation treatment has deleterious effects on neoangiogenesis and can delay the vascularization of the biomaterial (Kesler et al. 2013; Mao 2006; Martin 2013; Reinhold et al. 1990).
The advantage of in vivo vascularized tissue engineered grafts is the formation of a native complex branched vascular network with physiologically functional vessels within the engineered implant. However, a major drawback of this method is that the neovascularization of the implanted matrix needs several days to weeks, depending on the size of the implant and the implantation site (Rouwkema et al. 2008). Hypoxic areas within an implant may have negative impact on the cell survival and proliferation after implantation (Vitacolonna et al. 2015b). Several strategies have been investigated in order to enhance the in vivo vascularization of an engineered graft (Phelps and Garcia 2010). A promising approach is the co-cultivation of adult stem cells with stromal and perivascular cells inside a matrix (Baiguera and Ribatti 2013; Baldwin et al. 2014; Kirkpatrick et al. 2011; Phelps and Garcia 2010; Schumann et al. 2014; Shepherd et al. 2006). Numerous wound healing studies demonstrated that adult stem cells are capable to improve significantly chronic and acute wound healing (Parekkadan and Milwid 2010). The currently most widely studied adult stem cells for the reconstruction of musculoskeletal structures are mesenchymal stem cells (MSC) (Bianco et al. 2001; Maxson et al. 2012). MSCs can differentiate into a variety of different cells of the mesoderm (Farini et al. 2014; Vater et al. 2011) and are able to induce indirect trophic effects by secretion of a large number of different growth factors and cytokines (Phinney and Prockop 2007). Endothelial progenitor cells (EPC) participate in vivo in the renewal of the vasculature and promote de-novo vessel formation (adult vasculogenesis), their therapeutic potential for revascularization of ischemic tissue or tissue-engineered implants have been intensively studied (Asahara et al. 1997; Zammaretti and Zisch 2005). Perivascular or mural cells like pericytes are an additional essential cell type in vascular formation. They regulate the maturation and stabilization of new formed vessels (Abramsson et al. 2002; Alajati et al. 2008; Bryan and D’Amore 2008; Hegen et al. 2011; Rouwkema et al. 2006). Stromal cells like fibroblasts play also an important role in wound healing by autocrine and paracrine secretion of growth factors and cytokines such as e.g. TGF-ß, FGF and VEGF (Costa-Almeida et al. 2015; Novaes et al. 2007; Wenger et al. 2005).
To investigate whether MSC and EPC co-seeded with autologous pericytes and fibroblasts are capable to accelerate the vascularization of a human acellular dermis in radiated and non-irradiated tissues in vivo, we established a dorsal skinfold chamber model in rats. The vessel ingrowth was examined in vivo using intravital microscopy at day 0, 3, 9 and 12. Unseeded matrices and matrices seeded with fibroblasts and pericytes served as control groups.
Materials and methods
Animals
Forty-eight male Fisher-344 rats (150 g, Charles-River, Germany) were used for this study. All animals were held in the vivarium of the University Medical Centre Mannheim (Study approval by state authorities: Regierungspräsidium Karlsruhe, Germany: AZ 35-9185.81/G-187/09).
Study design
The study comprised a total of 6 treatment groups and 2 control groups. The rats were divided initially into 2 main groups (n = 24 rats) of equal size. One main group received neoadjuvant irradiation prior to the implantation of the skinfold chamber, the other does not. Each of the two main groups were subdivided into four groups each with n = 6 rats. Group 1 and 2 (control groups 1) received dermis without cells. Animals in group 3 and 4 (control groups 2) received a fibroblast/pericytes augmented-dermis to assess the effects of the fibroblasts/pericyte co-culture on the neovascularization. Rats in group 5 and 6 received dermis seeded with fibroblasts, pericytes and MSCs in co-culture. Rats in group 7 and 8 received dermis with EPCs in co-culture with fibroblasts and pericytes. The matrices were microscopically examined at day 3, 6, 9 and 12 after transplantation. The rats were sacrificed thereafter. Table 1 details the study design.
Human acellular dermis
Epiflex® (German Institute for Cell and Tissue Replacement (DIZG), Berlin, Germany) was used as human acellular dermis (hAD). The mechanical processing, decellularization, sterilization, preservation methods (Rossner et al. 2011; Vitacolonna et al. 2014) and composition (Roessner et al. 2012) are described in detail elsewhere.
Isolation and culture conditions of fibroblasts from rat subcutaneous fat
Autologous fibroblasts were obtained from the subcutaneous fat as described previously (Vitacolonna et al. 2015b). Briefly, the adipose tissue was digested using 2 mg/ml collagenase type 2 at 37 °C for 2 h. The suspension was washed twice with Dulbecco’s modified Eagle medium (DMEM). After digestion, the suspension was centrifuged at 400 g for 5 min. The resultant cell pellet was plated onto 100 mm2 tissue culture plates (Greiner Bio One, Germany) supplemented with DMEM (with 10% FBS and 1% penicillin/streptavidin solution (PAA, Germany)) and maintained at 37 °C in an incubator with 5% CO2.
Isolation and culture conditions of primary endothelial progenitor cells (EPC) from rat bone marrow
EPCs were isolated from tibia and femur of fischer-344 rats as described modified elsewhere (Kahler et al. 2007). Bone marrow was flushed out with DMEM supplemented with 10% FBS, 1000 U/ml Heparin (Sigma-Aldrich, USA) and 100U/ml penicillin G and 100 mg/ml streptomycin using a 20G needle (Becton–Dickinson, USA). To remove bone fragments and to obtain a single cell suspension, bone marrow was filtered through a 100 µm cell strainer (Becton–Dickinson, USA). The mononuclear cell fraction was obtained by a density gradient centrifugation using HSITOPAQUE®-1083 (Sigma-Aldrich, USA) for 30 min at 400 g. The mononuclear cell fraction was transferred into a new 50 ml tube, washed 3× with 30 ml DMEM and centrifuged at 250 g for 10 min. The resultant cell pellet was then suspended in EBM2-MV Medium (Lonza, USA) supplemented with 5% FBS and 100 U/ml penicillin G and 100 mg/ml streptomycin. Cells were plated on 6-well plates coated with 5 µg/cm2 rat-derived fibronectin (Sigma-Aldrich, USA), 10 µg/cm2 collagen I (Sigma-Aldrich, USA) and 2 µg/cm2 laminin (Sigma-Aldrich, USA). After 24 h the non-adherent cell population was transferred to new coated wells. The cells were cultured for further 24 h. This procedure was repeated to remove rapidly adherent cells such as mature endothelial cells or MSCs. The resultant fraction was cultured in EBM-2-MV medium containing vascular endothelial growth factor (VEGF), human fibroblast growth factor-B (hFGF-B), insulin like growth factor (IGF-1), human epidermal growth factor (hEGF), stem cell growth factor (SCGF), ascorbic acid, hydrocortisone, gentamycin and amphotericin B (MV-Kit, Lonza, USA). In addition, rat-specific rFGF and rVEGF (both R&D Systems, USA) were supplemented at a concentration of 100 ng/ml each. Medium was changed every 3 days. After 20 days of cultivation, cells were characterized by immunofluorescence staining and flow cytometry.
Characterization of EPCs from rat bone marrow
EPCs were characterized by immunofluorescence, flow cytometry and the incorporation of acetylated low density lipoprotein (acLDL; Biomedical Technologies, USA). Immunofluorescence staining was performed using antibodies against rat CD11b-biotin (AbD Serotec, USA), CD31 (Genetex, USA), CD34-AlexaFluor488 (Santa Cruz Biotechnology, Inc., USA), CD45-biotin (Cederlane, USA), CD106 (Becton–Dickinson, USA), CD133 (Abnova, USA), FLK-1 (Santa Cruz, USA) and Von Willebrand factor (vWF) (Santa Cruz, USA). Secondary FITC-labeled antibodies goat anti-mouse-IgG, goat anti-rabbit-IgG and streptavidin were purchased from Rockland (Rockland Immunochemicals, USA). Cells were fixed with ice cold acetone for 10 min. Stainings were performed in 8-well culture slides (BD Falcon™, Becton–Dickinson, USA) using manufacturers protocol. After blocking for 20 min with PBS containing 1% BSA (PAA, Germany), cells were incubated with primary antibodies for 2 h at room temperature (RT). After incubation, cells were washed 3 times with PBS for 5 min and incubated with secondary antibodies for 1 h. After 3 subsequent washing steps with PBS for 5 min VECTASHIELD mounting medium with DAPI (Vector Labs, USA) was used to prevent fading. Staining with acLDL was performed using manufacturer’s protocol. For additional characterization, flow cytometry (FACS) analyses were performed using standard protocol. Antibodies against rat CD11b-biotin (AbD Serotec, USA), CD31 (Genetex, USA), CD34-AlexaFluor488 (Santa Cruz, USA), CD44-Fitc (Immunotools, USA), CD45-biotin (Cederlane, USA), CD54-Biotin (Cederlane, USA), CD73 (Becton–Dickinson, USA), CD106 (Becton–Dickinson, USA), CD133 (Abnova, USA) and FLK-1 (Santa Cruz, USA) were used. Secondary FITC-labeled antibodies goat anti-mouse IgG, goat anti-rabbit IgG and streptavidin were purchased from Rockland (Rockland Immunochemicals, USA). Briefly, 1x106 cells/ml were incubated for 30 min at 4 °C with the primary antibodies. After three washing steps with PBS supplemented with 1% FBS, cells were incubated for further 30 min at 4 °C with the secondary antibodies. Measurements were done with a FACScalibur cytometer equipped with a 488 nm argon laser (Becton–Dickinson, USA) using BD CellQuest software. At least 10,000 events were collected and WinMDI 2.8 software was used to create the histograms.
Isolation and culture conditions of primary mesenchymal stem cells (MSC) from rat bone marrow
MSC were isolated from tibia and femur of fischer-344 rats based on the plastic adherence of these cells (Schumann et al. 2009). Subsequently, an enrichment using the surface marker CD90 as described elsewhere (Zhang and Chan 2010) was performed in order to increase the purity of the MSCs. Bone marrow was isolated and processed as described in the EPC section. After density gradient centrifugation, the mononuclear cells were plated in 10 cm dishes and cultured overnight in DMEM supplemented with 10% FBS, 100U/ml penicillin G, 100 mg/ml streptomycin and 50 ng/ml rat fibroblast growth factor (bFGF) (R&D Systems, Germany). After 24 h, the supernatant was aspirated and 10 ml of fresh medium was added. The cells were incubated for 1 week at 37 °C in a 5% CO2 incubator and the medium was changed every 3 days. Once the plates were approximately 80% confluent, the purity of the MSCs was increased by a positive isolation using CD90-biotin (Cederlane, USA) and streptavidin-coupled Dynabeads (Dynabeads® system; Invitrogen-Dynal, Sweden). The purification was performed according to the manufacturer’s instructions. Briefly, 1 × 107 cells/ml were incubated with CD90-biotin for 10 min at 4 °C. After 3 washing steps, cells were incubated for 20 min at 4 °C with the streptavidin-beads and washed afterwards three times. After the magnetic isolation procedure, cells were resuspended in DMEM supplemented with 10% FBS and 50 ng/ml rbFGF and plated at a density of 500cells/cm2 in 10 cm dishes. The cells were incubated at 37 °C in a 5% CO2 incubator and the medium was changed every 3 days. Once the cells were approximately 80% confluent, they were trypsinized and diluted to a concentration of 500cells/cm2 in 10 cm dishes.
Characterization of MSC from rat bone marrow
For immunofluorescence characterization, cells were stained with rat anti CD29-biotin (Becton–Dickinson, USA), CD90-biotin (Cederlane, USA), CD73 (Becton–Dickinson, USA), CD44-Fitc (Immunotools, USA), CD34-AlexaFluor-488 (Santa Cruz, USA), CD45-biotin (Cederlane, USA), CD31 (Genetex, USA), CD106 (Becton–Dickinson, USA), vWF (Santa Cruz, USA), fibronectin (Abcam, USA) and collagen I (Rockland, USA). Secondary FITC-labeled antibodies goat anti-mouse-IgG, goat anti-rabbit-IgG and streptavidin were purchased from Rockland (Rockland Immunochemicals, USA). Flow cytometry analyses were performed as described above. Antibodies against rat anti CD29-biotin, CD90-biotin, CD73, CD44-Fitc, CD34-AlexaFluor-488, CD45-biotin, CD31, CD106, fibronectin and collagen I were used.
To proof the preservation of multi-potency, two different differentiation assays were performed following the manufacturers protocol. For the differentiation of MSC into osteoblasts, cells were cultured 21 days in OsteoPrime® Induction Medium (Invitrogen-Gibco, USA) supplemented with ascorbic acid 2-phosphate, ß-glycerophosphate and dexamethasone. To show calcium deposition, cultures were washed once with PBS and stained for 5 min at RT with Alizarin Red S (Sigma-Aldrich, USA). To induce adipogenic differentiation, MSCs were cultured for 21 days in StemPro® Adipogenesis Differentiation Kit (Invitrogen-Gibco, USA). To stain the adipocytes, cells were fixed in 10% paraformaldehyde (Merck, Germany) for 1 h at RT and subsequently stained 20 min at RT with Oil-Red-O (3 volumes of 0.5% Oil-Red-O in isopropanol added to 2 volumes of aqua dest) (Sigma-Aldrich, USA).
Isolation and culture conditions of primary pericytes from rat bone marrow
Pericytes were isolated from tibia and femur of fischer-344 rats as described above in the EPC section. After density gradient centrifugation, depletion of MSC by negative magnetic separation with the MSC marker CD73-biotin (Becton–Dickinson, USA) was carried out as described above. The resultant cell pellet was plated in 10 cm dishes and cultured in EBM2-MV medium supplemented with 5% FBS, VEGF, hFGF-B, IGF-1, hEGF, SCGF, ascorbic acid, hydrocortisone, gentamycin and amphotericin B (MV-Kit, Lonza, USA). In addition, rat-specific rFGF and rVEGF (both R&D Systems, USA) were supplemented at a concentration of 100 ng/ml each. After about 10–15 days, isolated colonies were identified with different morphologies, containing presumably mature endothelial cells, EPC and MSC. Pericytes were identified based on their typical round nucleolus and numerous cell extensions as described in the literature (Bryan and D’Amore 2008; Dore-Duffy and Cleary 2011; Tigges et al. 2012). To purify the pericytes, isolated colonies were trypsinized using cloning rings (Sigma-Aldrich, USA). The detached cells were pooled and transferred into a collagen I, fibronectin, and laminin-coated 6-well plate. This procedure was repeated 2-4 times in order to obtain a homogeneous pericyte culture. EBM2-MV medium was changed every 3 days. After 20 days of cultivation, cells were characterized by immunofluorescence staining.
Characterization of primary pericytes from rat bone marrow
For immunofluorescence characterization, cells were stained with different markers typically expressed by pericytes (Farrington-Rock et al. 2004; Lamagna and Bergers 2006). Since there exists no specific marker for pericytes, antibodies against rat anti CD54 (Cederlane, USA), desmin (DAKO, Germany), smooth muscle actin (α-SMA) (DAKO, Germany) and platelet-derived growth factor receptor-ß (PDGFR-ß) (Becton–Dickinson, USA) were used. Secondary FITC-labeled antibody goat anti-mouse-IgG and streptavidin were purchased from Rockland (Rockland Immunochemicals, USA).
Irradiation
Twenty-four animals were assigned to the irradiation group. The radiation has been described previously (Vitacolonna et al. 2015b). Dorsal skin of the anesthetized animals was positioned under the irradiation apparatus and fixed using surgical clips. Twenty gray were administered topically 14 days prior to the implantation of the chamber in a single dose with an Intrabeam® device (PEC Photoelectronic Corporation PRS400; Voltage 50 kV, Current 40 µA; Run Time 6 min).
Implantation of the dorsal skinfold chamber
The surgical implantation of the skinfold chamber has been described previously in detail (Vitacolonna et al. 2015b). Briefly, after anesthesia the back skin was shaved, depilated and disinfected. Using an operation microscope (Zeiss OPMI 9-FC, Zeiss, Germany) an area (15 mm diameter) of the top layer of the dorsal was circularly excised and the underlying fascial layers were removed carefully from the thin skin muscle layer (panniculus carnosus). The remaining layer of the striated muscle, the subcutaneous tissue, the dermis and the epidermis, were covered with the counterpart of the dorsal skinfold chamber and screwed together. The observation window was filled with Ringer’s solution and covered with a detachable cover glass (Menzel, Germany). After preparation, the animals were allowed to recover from anesthesia and surgery for 48 h before the implantation of the scaffolds. During the period of study, the chambers and the implanted biomaterial underwent qualitative daily assessment. Animals with pathological findings or chamber defects were excluded from the experiment. Inflammation, hemorrhage, edema and persisting restriction of movement with consequent impairment of nourishment were termination criteria.
Cell seeded acellular dermis
Human acellular dermis (Epiflex®, DIZG, Berlin, Germany) with a thickness of 0.5–0.8 mm were cut into 5 mm × 5 mm pieces and rehydrated in DMEM for 2 h at 37 °C in 48-well plates. The rehydrated hADs were initially degassed with a chamber evacuation method to remove air trapped within the matrix (Hasegawa et al. 2010; Vitacolonna et al. 2013) and subsequently seeded with fibroblasts, pericytes and MSCs or EPCs in co-culture dynamically at a concentration of each 5x105 cells/hAD. Dynamic seeding (centrifugal cell immobilization) is described modified in a previous work (Vitacolonna et al. 2015a). Briefly, scaffolds were placed in a 48-well plate and a cell suspension with the above mentioned concentration was pipetted onto the scaffolds. The plate was then centrifuged at 300 g for 5 × 1 min in a common plate centrifuge, in order to allow a deeper penetration of the cells inside the dermis. Culture plates were incubated after the seeding procedure at 37 °C with 5% CO2 for 2 h to enable cell attachment. Immediately thereafter, the matrices were implanted into the skinfold chamber.
Implantation of the seeded dermis
The animals were anesthetized as described before. The cover slip was removed, the seeded graft was attached to the striated muscle in the centre of each chamber window with an 8/0 suture (Ethicon, USA). The window was closed with a sterile coverslip avoiding air bubbles.
In-situ fluorescence microscopy
Microscope examination was performed under anesthesia (as described above) at days 0, 3, 6, 9 and 12 using an Axiotech® Vario 100 intravital microscope (Zeiss, Germany). The 4 borders of the implanted dermis were defined as regions of interest (ROIs). Five pictures were taken from each ROI using a 5× objective (each ROI corresponded an area of 5 mm × 1 mm). The images were captured with a digital camera (AxioCam ICm1, Zeiss, Germany). Images were acquired with Axiovision LE V4.8.2 software (Zeiss, Germany). Images were stitched together using ICE (Image Composite Editor, V1.4.4.0, Microsoft, USA) to an edge-to-edge view of the full width of each ROI.
Quantification of vessel density
To quantify the vascularization of the implanted matrices, vessel density per ROI was analyzed. Whole photographs of each ROI (composed of each 5 single pictures) were binarized and segmented using FIJI and the advanced WIKA segmentation plug-in (Schindelin et al. 2012). Vessel density in the observed area was calculated automatically and expressed as % area occupied by blood vessels per ROI. A total of n = 24 ROIs per group were evaluated.
Statistics
All statistical tests were conducted with GraphPad Prism V6 (GraphPad Software Inc, USA). The data were analyzed by two-way ANOVA and a subsequent Bonferroni´s multiple comparisons test (<0.05 at a confidence level of 95%. p < 0.05 was considered significant).
Results
Characterization of EPC from rat bone marrow
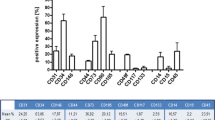
Figure 1a1–2 shows representative colonies of EPCs typically growing between 10 and 15 days in culture. To confirm the endothelial phenotype, the cells were stained after 20 days in culture for immunofluorescence characterization using specific antibodies and Dil-Ac-LDL. The cells were positive for Dil-Ac-LDL, CD11b, CD31, CD106, CD133, VEGFR2, vWF and CD34 (Fig. 1a3–10). The cells showed no expression of CD29, CD73 and the hematopoietic markers CD45 (data not shown). The cytometry analysis demonstrated the expression of CD11, CD31, VEGFR2, CD133, CD34 and CD54, but they were negative for CD29, CD73 und CD45 (Fig. 1b). The absence of CD29, CD45 and CD73 indicates that neither hematopoietic cells nor MSC or fibroblasts were present in the culture.

a1–2 EPC from bone marrow 10 and 20 days after isolation, cultured in EBM-2 medium; a3–10 Immunofluorescent characterization of the isolated EPC. Cells were positiv for Dil-AC-LDL, CD11b, CD31, CD34, CD106, CD133, FLK-1 and vWF. Cell nuclei were stained blue with DAPI. b The FACS analysis of the EPC illustrates the different degrees of expressions of the endothelial marker CD11b, CD31, VEGFR2, CD133, CD34 and CD54. The absence of CD29, CD73 and CD45 shows the high purity of the culture. Red represents the respective isotype control. (Color figure online)
Characterization of MSC from rat bone marrow
Adherent MSCs were present after 5–7 days in culture with spindle-shaped fibroblastoid morphology. The MSCs grew in colonies clearly defined from each other (Fig. 2a1–2). Immunofluorescence characterizations demonstrated expression of MSCs marker CD29, CD73, CD90, CD44, fibronectin and collagen-I (Fig. 2a3–8). Staining with CD14, CD45, CD31 and CD34 were negative (data not shown). Using flow cytometry, the cells expressed the stromal antibodies CD29, fibronectin, CD44, CD90 and CD73, whereas neither the macrophage marker CD14 nor CD31 and CD34 were expressed. The hematopoietic stem cell marker CD45 was weakly detected (<5%) (Fig. 2b). The majority of the MSCs cultured in osteogenic differentiation medium exhibited significant morphological changes including the production of a mineralized matrix, typically formed by osteoblasts (stained red with Alizarin Red) (Fig. 2c1). Cells incubated in adipogenic medium expressed lipid vacuoles formations of different sizes. The vacuoles were stained with the lipid dye Oil-Red-O to prove the differentiation into adipocytes (Fig. 2c2). Thus, accordingly to the International Society of Cellular Therapy (ISCT) the common set of minimal criteria for defining MSCs were confirmed in our study (Dominici et al. 2006).

a1–2 Phase contrast microscopic images of MSC colonies after 20 days of culture. a3–8 Immunofluorescence staining of MSC with antibodies against CD73, CD90, CD29, CD44, fibronectin and collagen-1. Cell nuclei were stained blue with DAPI. b FACS analysis of the MSC. The cells were positive for CD29, fibronectin, CD44, CD90, CD73 and negative for CD45, CD14, CD31 and CD34. Red represents the respective isotype control. c1 Cell culture in osteogenic differentiation medium led to cell differentiation into osteoblasts. Cells were stained with alizarin red to visualize the formed mineralized matrix. c2 Culture in adipogenic differentiation medium led to cell differentiation into adipocytes. The lipid droplets were stained by the lipid dye Oil-Red-O. (Color figure online)
Characterization of pericytes from rat bone marrow
After isolation of the pericytes using cloning rings, a relatively homogeneous population could be cultured for numerous passages (Fig. 3a, b). The characteristic prominent nucleus, irregular triangular cell bodies and long extensions of pericytes (Hirschi and D’Amore 1996; Liu et al. 2014) were present in all cells used in this experiment (Fig. 3b). All cells expressed desmin, α-SMA, CD54 and PDGFR-ß (Fig. 3c–f).

Phase contrast microscopic images of pericytes after a 7 and b 14 days in culture. c–f Immunofluorescent staining of pericytes were positiv for Desmin, α-SMA, CD54 and PDGFR-beta. Cell nuclei were stained blue with DAPI. (Color figure online)
Quantification of vessel densities
The vessel densities were analyzed semi-automatic using the WEKA segmentation plugin and FIJI and expressed as % vessel density per observed region. Group 1 and 2 (unseeded hAD; ±radiation) were compared with group 3 and 4 (hAD seeded with fibroblasts and pericytes; ±radiation) to assess the influence of the co-cultured fibroblasts and pericytes on dermis vascularization. The control groups 3 and 4 (hAD seeded with fibroblasts +pericytes; ±radiation) were compared with the stem cell-augmented matrices (group 5–8) to evaluate the influence of EPCs and MSCs on the vascularization.
Figure 4 shows representative intravital microscopic pictures of an observed ROI between day 6–12 (group 5: +fibroblasts +pericytes +MSC −radiation). Perfused macro-vessels were clearly distinguishable from the high intrinsic fluorescence of the dermis without fluorescent staining. In all groups first vessel ingrowth were observable at day 6 with an exception of group 5 (+fibroblasts +pericytes +MSC −radiation). In group 5 first vessel ingrowth became visible at day 3 within the border zones.

Representative intravital microscopic picture of the observed ROI between day 6 to 12 (group 5: hAD +fibroblasts/pericytes +MSC −radiation). Perfused macro-vessels were clearly distinguishable from the high intrinsic fluorescence of the dermis. Vessel ingrowth started to be visible at the borders of the hAD. Newly formed macro-vessels interconnect with each other and form a dense microvascular network within the border zones until day 12
At any time point the macrovascular density of cell seeded scaffolds was significantly higher compared to the unseeded control groups with an exception at day 12 in the non-irradiated groups. In addition, we found, that the irradiation caused a significant decrease of vessel density in all groups compared to the non-irradiated groups during the entire observation.
Figure 5 displays the calculated vessel densities of the non-irradiated groups (group 1, 3, 5 and 7).

Vessel densities in %/ROI of the non-irradiated groups at day 3–12
The comparison between the control group 1 (group 1; −cells −rad) and control group 2 (group 3; +fibroblasts +pericytes −rad) showed no differences at day 3 (both groups 0%). At day 6 the vessel density in group 3 was significant higher than in group 1 (group 1: 3.6 ± 0.4% and group 3: 16.2 ± 1.3%; p = 0.0217). At day 9 a significant higher vessel ingrowth was detectable in the seeded hAD (group 3: 35.6 ± 4.6%) compared to the unseeded hAD (group 1: 14.7 ± 5.3%) (p < 0.0001). At day 12 there were no differences between both non-irradiated control groups (group 1: 28.4 ± 5.4% and group 3: 39.2 ± 19.5%).
Co-seeding with MSCs (group 5) leads to an earlier detectable vessel ingrowth starting at day 3 (3.1 ± 2.2%) compared to the other groups. The EPC group (group 7) achieved none visible vascularization at day 3. At day 6 vessel density in group 5 (23.9 ± 1.9%) was significant higher than in group 3 (p = 0.0324). Group 7 (+EPC) showed no differences (14.2 ± 2.2%) compared to the control group 2. At day 9 vessel density in group 5 (+MSC) was significant higher (47.6 ± 2.1%) than in group 3 (p = 0.0327) while group 7 (+EPC) showed no significant difference (38.0 ± 4.5%). At day 12 neither MSCs nor EPCs increased the vessel density compared to group 3 (group 5: 46.6 ± 13.9% and group 7: 41.1 ± 24.1%).
Figure 6 depicts the vessel densities from the irradiated groups 2, 4, 6 and 8.

Vessel densities in %/ROI of the irradiated groups at day 3–12
The comparison at day 3 between control group 1 (group 2; −cells +rad) and control group 2 (group 4; +fibroblasts +pericytes +rad) showed in both groups none visible vessel ingrowth. At day 6 vessel ingrowth was not detectable in the unseeded matrices (group 2) while matrices of control group 2 (group 4; +fibroblasts +pericytes +rad) achieved a vessel density of 3.1 ± 0.4% per ROI (p = 0.0004). At day 9 vessel density in group 2 (2.2 ± 1.3%) was significant lower than in group 4 (8.4 ± 0.7%) (p = 0.0008). At day 12 vascular density in group 4 (12.9 ± 5.2%) increase compared to the non-seeded matrices (group 2; 4.3 ± 3.1%) (p < 0.0001).
In none of the stem cell-augmented groups (group 6 and group 8) and control group 2 (group 4) were vessels detectable at day 3. At day 6 matrices co-seeded with MSCs (group 6) achieved a higher vessel density (9.6 ± 1.2%) than control group 2 (group 4; p = 0.0004). EPCs (group 8; 2.5 ± 0.8%) had no beneficial effect on dermis vascularization compared to the fibroblasts +pericytes-seeded control group. MSCs showed also at day 9 a superior vessel growth (21.8 ± 1.2%) in comparison to group 4 (p < 0.0001). Even at day 9 EPCs were not able to increase vascular density significantly (group 8; 6.3 ± 1.5%). At day 12 the co-seeding of MSCs lead to a higher vessel density (group 6; 22.4 ± 10.0%) (p < 0.0001). As well as seen at earlier time points no improvement of vascularization could be detected in the EPC group 8 (11.0 ± 3.7%).
Discussion
In the present study we compared the vascularization potential of EPC and MSC in co-culture with fibroblasts and pericytes seeded on an acellular human dermis. We were further simulating a multi-modal neoadjuvant therapeutic approach by pre-implantation irradiation to investigate the effect of radiation on the vessel ingrowth.
Our study demonstrates that the pre-irradiation treatment results in a significant weaker graft vascularization in all groups. The augmentation with fibroblasts and pericytes in co-culture significantly enhanced the vascularization at the border zones of the matrices compared to the unseeded matrices in both, irradiated and non-irradiated groups. These results are consistent with previous findings showing that vitalization of scaffolds with cells causes a significant improvement in blood vessel formation in vivo (Kampmann et al. 2013; Laschke et al. 2013; McFadden et al. 2013; Schumann et al. 2014). In many wound healing studies the transplantation of vital, non-irradiated fibroblasts resulted in normal wound healing or an improvement of wound breaking strength after radiation (Chen et al. 2010; Dantzer et al. 2003; Ferguson et al. 1999; Roessner et al. 2013, 2011). Fibroblasts act as important indirect supporting cells during angiogenesis by constitutively expressing angiogenic factors, which regulate endothelial tube formation (Baldwin et al. 2014). As well, perivascular cells like pericytes provide direct support to endothelial cells and are essential to stabilizing and maturing new vessels and capillaries in tissue engineered implants (Golas et al. 2013; Jain 2003; Stratman and Davis 2012; Yamamoto et al. 2010; Yang et al. 2012).
The co-seeding of MSCs leads into a further significant increase of vessel density. Their regenerative effects seem to induce the vascular growth in hypoxic tissue in both non-irradiated and irradiated tissues. MSCs demonstrated the capability to improve wound healing in both animal models and humans in the treatment of acute and chronic wounds (Falanga et al. 2007; Kwon et al. 2008; Liu et al. 2006; McFarlin et al. 2006; Shin and Peterson 2013; Wu et al. 2007; Yoshikawa et al. 2008). The reparative and angiogenic properties of MSCs were also intensively studied in the recent years (Au et al. 2008; Hegen et al. 2011; Sanz et al. 2008). For example, MSC were used to engraft and ameliorate limb ischemia or to enhance angiogenesis in different stroke models (da Cunha et al. 2013; Hoffmann et al. 2010; Honmou et al. 2012; Madonna et al. 2013; Martins et al. 2014; Watt et al. 2013; Wu et al. 2007). Recent studies indicate that the involvement of MSCs in wound healing is not limited by engraftment and differentiation, but also by broad immunoregulatory effects, possessing immunosuppressive and anti-inflammatory properties (Baiguera et al. 2012; Bernardo and Fibbe 2013; English 2013; Salem and Thiemermann 2010; Yagi et al. 2010). In addition, MSCs secret a variety of bioactive factors that have angiogenic and antiapoptotic properties and serve to limit tissue damages at the injured sites, regenerating blood supply, and recruiting progenitor cells (Murray et al. 2014). MSCs have as well been shown to be an effective therapy option in the treatment of acute radiation injuries by enhancing the recovery of hematopoiesis and reducing apoptosis (Chang et al. 2013; Hu et al. 2010; Lange et al. 2011; Semont et al. 2006). Although we have demonstrated a significant increase in the vascularization in the MSC groups, the exact mechanism behind is still unclear. However, we hypothesize that a multifactorial interaction of their varied properties may be responsible for this effect. To elucidate the exact mechanism, further studies are needed.
The co-seeding with EPCs had no effect on the vessel density, although they were used in numerous studies to improve vascularization of wounds after transplantation or injection in vivo (Hess et al. 2002; Kocher et al. 2001; Takahashi et al. 1999). In contrast to these results, there are some studies, in which transplanted EPCs caused an aggravation of the disease or injury (Pearson 2010). EPCs consist of at least two subtypes, the “early”—and “late”-outgrowth EPCs with different morphologies and growth patterns. Late-EPCs exhibit a high proliferative capacity and typical endothelial antigens (Mukai et al. 2008). It was reported, that late-EPCs augment the angiogenesis by direct incorporation into the neovasculature and the secretion of angiogenic growth factors, while early-EPCs are very short-lived in culture and not able to form tubular structures (Hur et al. 2004; Suh et al. 2005). Instead, it was postulated that these cells are involved rather in inflammatory processes than in angiogenesis (Fadini et al. 2008). The EPCs used in this study were derived from the non-adherent fraction of mononuclear bone marrow cells, which grew after 48 h in fibronectin-coated cell culture plates to angioblast-shaped cells as reported before (Salazar et al. 2001). The cells presented within the first 5 days in culture an elongated, thin spindle-shaped morphology, as described in the literature as “early-outgrowth” EPCs. Between day 8–15 the proliferation rate increased and the cells showed an endothelial cell-like shape, which may be typical for “late outgrowth” EPCs. FACS analysis of the transplanted EPCs demonstrated a weak expression of the monocyte marker CD14 after 15 days in culture. After more than 2 weeks in culture, the “late”—subtype dominated. These cells expressed typical antigens of endothelial origin, such as CD34, CD31, or vWF. Although we have seeded late-subtype EPCs onto the hADs, they did not increase the vessel density compared to the groups with fibroblast/pericyte-seeded matrices. The results suggest that the in vitro expansion decreased the migratory capacity of the EPCs and led to a maturation of the cells into endothelial cells which resulted into a partial loss of their vasculogenic ability, as reported elsewhere (Ingram et al. 2004; Khan et al. 2006; Melero-Martin et al. 2007; Singh et al. 2011).
Limitation of the study
The unequivocal verification of purity of the pericyte culture with antibodies against desmin, SMA, PDGFR-ß and CD54 may be critical due to the lack of unique pericyte-specific markers. So far, known pericyte markers are also expressed in various other cell types (Kloc et al. 2015). Therefore, the identification may rely on multiple criteria including morphology and co-expression of several different pericyte markers.
Conclusion
We demonstrated that the co-seeding of fibroblasts, pericytes and MSCs have the potential to accelerate the vascularization of an implanted matrix. Since MSCs are easy to isolate, possess an extraordinary plasticity and retain their ability to differentiate even after massive in vitro expansion, their co-seeding with target cells into a graft prior the surgical intervention could represent a promising vascularization strategy to increase the vessel number within the construct in vivo after implantation.
References
Abramsson A, Berlin O, Papayan H, Paulin D, Shani M, Betsholtz C (2002) Analysis of mural cell recruitment to tumor vessels. Circulation 105:112–117
Alajati A et al (2008) Spheroid-based engineering of a human vasculature in mice. Nat Methods 5:439–445. doi:10.1038/nmeth.1198
Asahara T et al (1997) Isolation of putative progenitor endothelial cells for angiogenesis. Science 275:964–967
Au P, Tam J, Fukumura D, Jain RK (2008) Bone marrow-derived mesenchymal stem cells facilitate engineering of long-lasting functional vasculature. Blood 111:4551–4558. doi:10.1182/blood-2007-10-118273
Baiguera S, Ribatti D (2013) Endothelialization approaches for viable engineered tissues. Angiogenesis 16:1–14. doi:10.1007/s10456-012-9307-8
Baiguera S, Jungebluth P, Mazzanti B, Macchiarini P (2012) Mesenchymal stromal cells for tissue-engineered tissue and organ replacements Transplant international. Transplantation 25:369–382. doi:10.1111/j.1432-2277.2011.01426.x
Baldwin J et al (2014) In vitro pre-vascularisation of tissue-engineered constructs A co-culture perspective. Vasc Cell 6:13. doi:10.1186/2045-824X-6-13
Bernardo ME, Fibbe WE (2013) Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell 13:392–402. doi:10.1016/j.stem.2013.09.006
Bianco P, Riminucci M, Gronthos S, Robey PG (2001) Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells 19:180–192. doi:10.1634/stemcells.19-3-180
Bryan BA, D’Amore PA (2008) Pericyte isolation and use in endothelial/pericyte coculture models. Methods Enzymol 443:315–331. doi:10.1016/S0076-6879(08)02016-8
Chang P, Qu Y, Liu Y, Cui S, Zhu D, Wang H, Jin X (2013) Multi-therapeutic effects of human adipose-derived mesenchymal stem cells on radiation-induced intestinal injury. Cell Death Dis 4:e685. doi:10.1038/cddis.2013.178
Chen X, Aledia AS, Popson SA, Him L, Hughes CC, George SC (2010) Rapid anastomosis of endothelial progenitor cell-derived vessels with host vasculature is promoted by a high density of cotransplanted fibroblasts. Tissue Eng Part A 16:585–594. doi:10.1089/ten.TEA.2009.0491
Choi JH et al (2010) Adipose tissue engineering for soft tissue regeneration. Tissue Eng Part B Rev 16:413–426. doi:10.1089/ten.TEB.2009.0544
Costa-Almeida R, Gomez-Lazaro M, Ramalho C, Granja PL, Soares R, Guerreiro SG (2015) Fibroblast-endothelial partners for vascularization strategies in tissue engineering. Tissue Eng Part A 21:1055–1065. doi:10.1089/ten.TEA.2014.0443
da Cunha FF, Martins L, Martin PK, Stilhano RS, Paredes Gamero EJ, Han SW (2013) Comparison of treatments of peripheral arterial disease with mesenchymal stromal cells and mesenchymal stromal cells modified with granulocyte and macrophage colony-stimulating factor. Cytotherapy 15:820–829. doi:10.1016/j.jcyt.2013.02.014
Dantzer D et al (2003) Effect of radiation and cell implantation on wound healing in a rat model. J Surg Oncol 83:185–190. doi:10.1002/jso.10242
Dominici M et al (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317. doi:10.1080/14653240600855905
Dore-Duffy P, Cleary K (2011) Morphology and properties of pericytes. Methods Mol Biol 686:49–68. doi:10.1007/978-1-60761-938-3_2
English K (2013) Mechanisms of mesenchymal stromal cell immunomodulation. Immunol Cell Biol 91:19–26. doi:10.1038/icb.2012.56
Fadini GP, Baesso I, Albiero M, Sartore S, Agostini C, Avogaro A (2008) Technical notes on endothelial progenitor cells: ways to escape from the knowledge plateau. Atherosclerosis 197:496–503. doi:10.1016/j.atherosclerosis.2007.12.039
Falanga V et al (2007) Autologous bone marrow-derived cultured mesenchymal stem cells delivered in a fibrin spray accelerate healing in murine and human cutaneous wounds. Tissue Eng 13:1299–1312. doi:10.1089/ten.2006.0278
Farini A, Sitzia C, Erratico S, Meregalli M, Torrente Y (2014) Clinical applications of mesenchymal stem cells in chronic diseases. Stem Cells Int 2014:306573. doi:10.1155/2014/306573
Farrington-Rock C, Crofts NJ, Doherty MJ, Ashton BA, Griffin-Jones C, Canfield AE (2004) Chondrogenic and adipogenic potential of microvascular pericytes. Circulation 110:2226–2232. doi:10.1161/01.CIR.0000144457.55518.E5
Ferguson PC, Boynton EL, Wunder JS, Hill RP, O’Sullivan B, Sandhu JS, Bell RS (1999) Intradermal injection of autologous dermal fibroblasts improves wound healing in irradiated skin. J Surg Res 85:331–338. doi:10.1006/jsre.1999.5664
Golas AR, Perez JL, Fullerton N, Lekic N, Hooper RC, Spector JA (2013) Vascular smooth muscle cell optimization of vasculogenesis within naturally derived, biodegradable, hybrid hydrogel scaffolds. Plast Reconstr Surg 132:952e–963e. doi:10.1097/PRS.0b013e3182a805df
Hasegawa T et al (2010) Efficient cell-seeding into scaffolds improves bone formation. J Dent Res 89:854–859. doi:10.1177/0022034510370022
Hegen A et al (2011) Efficient in vivo vascularization of tissue-engineering scaffolds. J Tissue Eng Regen Med 5:e52–e62. doi:10.1002/term.336
Hess DC, Hill WD, Martin-Studdard A, Carroll J, Brailer J, Carothers J (2002) Bone marrow as a source of endothelial cells and NeuN-expressing cells after stroke. Stroke 33:1362–1368
Hirschi KK, D’Amore PA (1996) Pericytes in the microvasculature. Cardiovasc Res 32:687–698
Hoffmann J, Glassford AJ, Doyle TC, Robbins RC, Schrepfer S, Pelletier MP (2010) Angiogenic effects despite limited cell survival of bone marrow-derived mesenchymal stem cells under ischemia. Thoracic Cardiovasc Surg 58:136–142. doi:10.1055/s-0029-1240758
Honmou O, Onodera R, Sasaki M, Waxman SG, Kocsis JD (2012) Mesenchymal stem cells: therapeutic outlook for stroke. Trends Mol Med 18:292–297. doi:10.1016/j.molmed.2012.02.003
Hu KX, Sun QY, Guo M, Ai HS (2010) The radiation protection and therapy effects of mesenchymal stem cells in mice with acute radiation injury. Br J Radiol 83:52–58. doi:10.1259/bjr/61042310
Hur J et al (2004) Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler Thromb Vasc Biol 24:288–293. doi:10.1161/01.ATV.0000114236.77009.06
Ingram DA et al (2004) Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 104:2752–2760. doi:10.1182/blood-2004-04-1396
Jain RK (2003) Molecular regulation of vessel maturation. Nat Med 9:685–693. doi:10.1038/nm0603-685
Kahler CM et al (2007) Peripheral infusion of rat bone marrow derived endothelial progenitor cells leads to homing in acute lung injury. Respir Res 8:50. doi:10.1186/1465-9921-8-50
Kampmann A et al (2013) Additive effect of mesenchymal stem cells and VEGF to vascularization of PLGA scaffolds. Microvasc Res 90:71–79. doi:10.1016/j.mvr.2013.07.006
Kesler CT et al (2013) Vascular endothelial growth factor-C enhances radiosensitivity of lymphatic endothelial cells. Angiogenesis. doi:10.1007/s10456-013-9400-7
Khan ZA, Melero-Martin JM, Wu X, Paruchuri S, Boscolo E, Mulliken JB, Bischoff J (2006) Endothelial progenitor cells from infantile hemangioma and umbilical cord blood display unique cellular responses to endostatin. Blood 108:915–921. doi:10.1182/blood-2006-03-006478
Kirkpatrick CJ, Fuchs S, Unger RE (2011) Co-culture systems for vascularization–learning from nature. Adv Drug Deliv Rev 63:291–299. doi:10.1016/j.addr.2011.01.009
Kloc M, Kubiak JZ, Li XC, Ghobrial RM (2015) Pericytes, microvasular dysfunction, and chronic rejection. Transplantation 99:658–667. doi:10.1097/TP.0000000000000648
Kocher AA et al (2001) Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med 7:430–436. doi:10.1038/86498
Krauss MC (1999) Recent advances in soft tissue augmentation. Semin Cutan Med Surg 18:119–128
Kwon DS et al (2008) Treatment with bone marrow-derived stromal cells accelerates wound healing in diabetic rats. Int Wound J 5:453–463. doi:10.1111/j.1742-481X.2007.00408.x
Lamagna C, Bergers G (2006) The bone marrow constitutes a reservoir of pericyte progenitors. J Leukoc Biol 80:677–681. doi:10.1189/jlb.0506309
Lange C et al (2011) Radiation rescue: mesenchymal stromal cells protect from lethal irradiation. PLOS ONE 6:e14486. doi:10.1371/journal.pone.0014486
Laschke MW et al (2006) Angiogenesis in tissue engineering: breathing life into constructed tissue substitutes. Tissue Eng 12:2093–2104. doi:10.1089/ten.2006.12.2093
Laschke MW et al (2013) Three-dimensional spheroids of adipose-derived mesenchymal stem cells are potent initiators of blood vessel formation in porous polyurethane scaffolds. Acta Biomater 9:6876–6884. doi:10.1016/j.actbio.2013.02.013
Liu Y, Dulchavsky DS, Gao X, Kwon D, Chopp M, Dulchavsky S, Gautam SC (2006) Wound repair by bone marrow stromal cells through growth factor production. J Surg Res 136:336–341. doi:10.1016/j.jss.2006.07.037
Liu G et al (2014) Isolation, purification, and cultivation of primary retinal microvascular pericytes: a novel model using rats. Microcirculation 21:478–489. doi:10.1111/micc.12121
Madonna R, Taylor DA, Geng YJ, De Caterina R, Shelat H, Perin EC, Willerson JT (2013) Transplantation of mesenchymal cells rejuvenated by the overexpression of telomerase and myocardin promotes revascularization and tissue repair in a murine model of hindlimb ischemia. Circ Res 113:902–914. doi:10.1161/CIRCRESAHA.113.301690
Mao XW (2006) A quantitative study of the effects of ionizing radiation on endothelial cells and capillary-like network formation. Technol Cancer Res Treat 5:127–134
Martin BJ (2013) Inhibiting vasculogenesis after radiation: a new paradigm to improve local control by radiotherapy. Semin Radiat Oncol 23:281–287. doi:10.1016/j.semradonc.2013.05.002
Martins L, Martin PK, Han SW (2014) Angiogenic properties of mesenchymal stem cells in a mouse model of limb ischemia. Methods Mol Biol 1213:147–169. doi:10.1007/978-1-4939-1453-1_13
Maxson S, Lopez EA, Yoo D, Danilkovitch-Miagkova A, Leroux MA (2012) Concise review: role of mesenchymal stem cells in wound repair. Stem Cells Transl Med 1:142–149. doi:10.5966/sctm.2011-0018
McFadden TM et al (2013) The delayed addition of human mesenchymal stem cells to pre-formed endothelial cell networks results in functional vascularization of a collagen-glycosaminoglycan scaffold in vivo. Acta Biomater 9:9303–9316. doi:10.1016/j.actbio.2013.08.014
McFarlin K et al (2006) Bone marrow-derived mesenchymal stromal cells accelerate wound healing in the rat. Wound Repair Regen 14:471–478. doi:10.1111/j.1743-6109.2006.00153.x
Melero-Martin JM, Khan ZA, Picard A, Wu X, Paruchuri S, Bischoff J (2007) In vivo vasculogenic potential of human blood-derived endothelial progenitor cells. Blood 109:4761–4768. doi:10.1182/blood-2006-12-062471
Mukai N et al (2008) A comparison of the tube forming potentials of early and late endothelial progenitor cells. Exp Cell Res 314:430–440. doi:10.1016/j.yexcr.2007.11.016
Murray IR et al (2014) Natural history of mesenchymal stem cells, from vessel walls to culture vessels. CMLS 71:1353–1374. doi:10.1007/s00018-013-1462-6
Novaes AB Jr, Marchesan JT, Macedo GO, Palioto DB (2007) Effect of in vitro gingival fibroblast seeding on the in vivo incorporation of acellular dermal matrix allografts in dogs. J Periodontol 78:296–303. doi:10.1902/jop.2007.060060
Parekkadan B, Milwid JM (2010) Mesenchymal stem cells as therapeutics. Annu Rev Biomed Eng 12:87–117. doi:10.1146/annurev-bioeng-070909-105309
Pearson JD (2010) Endothelial progenitor cells–an evolving story. Microvasc Res 79:162–168. doi:10.1016/j.mvr.2009.12.004
Phelps EA, Garcia AJ (2010) Engineering more than a cell: vascularization strategies in tissue engineering. Curr Opin Biotechnol 21:704–709. doi:10.1016/j.copbio.2010.06.005
Phinney DG, Prockop DJ (2007) Concise review: mesenchymal stem/multipotent stromal cells: the state of trans differentiation and modes of tissue repair–current views. Stem Cells 25:2896–2902. doi:10.1634/stemcells.2007-0637
Reinhold HS, Calvo W, Hopewell JW, van der Berg AP (1990) Development of blood vessel-related radiation damage in the fimbria of the central nervous system. Int J Radiat Oncol Biol Phys 18:37–42
Roessner ED, Thier S, Hohenberger P, Schwarz M, Pott P, Dinter D, Smith M (2011) Acellular dermal matrix seeded with autologous fibroblasts improves wound breaking strength in a rodent soft tissue damage model in neoadjuvant settings. J Biomater Appl 25:413–427. doi:10.1177/0885328209347961
Roessner ED, Vitacolonna M, Hohenberger P (2012) Confocal laser scanning microscopy evaluation of an acellular dermis tissue transplant (Epiflex(R)). PLOS ONE 7:e45991. doi:10.1371/journal.pone.0045991
Roessner E, Vitacolonna M, Schulmeister A, Pilz L, Tsagogiorgas C, Brockmann M, Hohenberger P (2013) Human acellular dermis seeded with autologous fibroblasts enhances bronchial anastomotic healing in an irradiated rodent sleeve resection model. Ann Surg Oncol 20(Suppl 3):S709–S715. doi:10.1245/s10434-013-3209-x
Rossner E et al (2011) Epiflex((R)) a new decellularised human skin tissue transplant: manufacture and properties. Cell Tissue Bank 12:209–217. doi:10.1007/s10561-010-9187-3
Rouwkema J, de Boer J, Van Blitterswijk CA (2006) Endothelial cells assemble into a 3-dimensional prevascular network in a bone tissue engineering construct. Tissue Eng 12:2685–2693. doi:10.1089/ten.2006.12.2685
Rouwkema J, Rivron NC, van Blitterswijk CA (2008) Vascularization in tissue engineering. Trends Biotechnol 26:434–441. doi:10.1016/j.tibtech.2008.04.009
Salazar AB, McAlister VC, Gupta R, MacDonald AS (2001) Circulating endothelial cells after transplantation. Lancet 357:1450
Salem HK, Thiemermann C (2010) Mesenchymal stromal cells: current understanding and clinical status. Stem Cells 28:585–596. doi:10.1002/stem.269
Sanz L et al (2008) Long-term in vivo imaging of human angiogenesis: critical role of bone marrow-derived mesenchymal stem cells for the generation of durable blood vessels. Microvasc Res 75:308–314. doi:10.1016/j.mvr.2007.11.007
Schindelin J et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi:10.1038/nmeth.2019
Schumann P et al (2009) Consequences of seeded cell type on vascularization of tissue engineering constructs in vivo. Microvasc Res 78:180–190. doi:10.1016/j.mvr.2009.06.003
Schumann P et al (2014) Accelerating the early angiogenesis of tissue engineering constructs in vivo by the use of stem cells cultured in matrigel. J Biomed Mater Res Part A 102:1652–1662. doi:10.1002/jbm.a.34826
Semont A et al (2006) Mesenchymal stem cells increase self-renewal of small intestinal epithelium and accelerate structural recovery after radiation injury. Adv Exp Med Biol 585:19–30
Shepherd BR, Enis DR, Wang F, Suarez Y, Pober JS, Schechner JS (2006) Vascularization and engraftment of a human skin substitute using circulating progenitor cell-derived endothelial cells. FASEB J 20:1739–1741. doi:10.1096/fj.05-5682fje
Shin L, Peterson DA (2013) Human mesenchymal stem cell grafts enhance normal and impaired wound healing by recruiting existing endogenous tissue stem/progenitor cells. Stem Cells Transl Med 2:33–42. doi:10.5966/sctm.2012-0041
Singh S, Wu BM, Dunn JC (2011) Accelerating vascularization in polycaprolactone scaffolds by endothelial progenitor cells. Tissue Eng Part A 17:1819–1830. doi:10.1089/ten.TEA.2010.0708
Stratman AN, Davis GE (2012) Endothelial cell-pericyte interactions stimulate basement membrane matrix assembly: influence on vascular tube remodeling, maturation, and stabilization. Microsc Microanal 18:68–80. doi:10.1017/S1431927611012402
Suh W et al (2005) Transplantation of endothelial progenitor cells accelerates dermal wound healing with increased recruitment of monocytes/macrophages and neovascularization. Stem Cells 23:1571–1578. doi:10.1634/stemcells.2004-0340
Takahashi T et al (1999) Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med 5:434–438. doi:10.1038/7434
Tigges U, Welser-Alves JV, Boroujerdi A, Milner R (2012) A novel and simple method for culturing pericytes from mouse brain. Microvasc Res 84:74–80. doi:10.1016/j.mvr.2012.03.008
Vater C, Kasten P, Stiehler M (2011) Culture media for the differentiation of mesenchymal stromal cells. Acta Biomater 7:463–477. doi:10.1016/j.actbio.2010.07.037
Vitacolonna M, Belharazem D, Hohenberger P, Roessner ED (2013) Effect of static seeding methods on the distribution of fibroblasts within human acellular dermis. Biomed Eng Online 12:55. doi:10.1186/1475-925X-12-55
Vitacolonna M et al (2014) Effect on the tensile strength of human acellular dermis (Epiflex(R)) of in vitro incubation simulating an open abdomen setting. BMC Surg 14:7. doi:10.1186/1471-2482-14-7
Vitacolonna M, Belharazem D, Hohenberger P, Roessner ED (2015a) Effect of dynamic seeding methods on the distribution of fibroblasts within human acellular dermis. Cell Tissue Bank. doi:10.1007/s10561-015-9508-7
Vitacolonna M, Belharazem D, Maier P, Hohenberger P, Roessner ED (2015b) In vivo quantification of the effects of radiation and presence of hair follicle pores on the proliferation of fibroblasts in an acellular human dermis in a dorsal skinfold chamber: relevance for tissue reconstruction following neoadjuvant therapy. PLOS ONE 10:e0125689. doi:10.1371/journal.pone.0125689
Watt SM, Gullo F, van der Garde M, Markeson D, Camicia R, Khoo CP, Zwaginga JJ (2013) The angiogenic properties of mesenchymal stem/stromal cells and their therapeutic potential. Br Med Bull 108:25–53. doi:10.1093/bmb/ldt031
Wenger A, Kowalewski N, Stahl A, Mehlhorn AT, Schmal H, Stark GB, Finkenzeller G (2005) Development and characterization of a spheroidal coculture model of endothelial cells and fibroblasts for improving angiogenesis in tissue engineering. Cells Tissues Organs 181:80–88. doi:10.1159/000091097
Wu Y, Chen L, Scott PG, Tredget EE (2007) Mesenchymal stem cells enhance wound healing through differentiation and angiogenesis. Stem Cells 25:2648–2659. doi:10.1634/stemcells.2007-0226
Yagi H, Soto-Gutierrez A, Parekkadan B, Kitagawa Y, Tompkins RG, Kobayashi N, Yarmush ML (2010) Mesenchymal stem cells: mechanisms of immunomodulation and homing. Cell Transpl 19:667–679. doi:10.3727/096368910X508762
Yamamoto M, James D, Li H, Butler J, Rafii S, Rabbany S (2010) Generation of stable co-cultures of vascular cells in a honeycomb alginate scaffold. Tissue Eng Part A 16:299–308. doi:10.1089/ten.TEA.2009.0010
Yang M, Chen CZ, Shu YS, Shi WP, Cheng SF, Gu YJ (2012) Preseeding of human vascular cells in decellularized bovine pericardium scaffold for tissue-engineered heart valve: an in vitro and in vivo feasibility study. J Biomed Mater Res Part B Appl Biomater 100:1654–1661. doi:10.1002/jbm.b.32734
Yoshikawa T et al (2008) Wound therapy by marrow mesenchymal cell transplantation. Plast Reconstr Surg 121:860–877. doi:10.1097/01.prs.0000299922.96006.24
Zammaretti P, Zisch AH (2005) Adult ‘endothelial progenitor cells’. Renewing vasculature. Int J Biochem Cell Biol 37:493–503. doi:10.1016/j.biocel.2004.06.018
Zhang L, Chan C (2010) Isolation and enrichment of rat mesenchymal stem cells (MSCs) and separation of single-colony derived MSCs. J Vis Exp. doi:10.3791/1852
Acknowledgements
The authors thank Dr. Jan C. Brune and Dr. Mark D. Smith from the German Institute for Cell and Tissue Replacement (DIZG) for advice relating to use of Epiflex® and for assistance with data analysis and editing of the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Mario Vitacolonna, Dr. Djeda Belharazem, Prof. Dr. Peter Hohenberger and Dr. Eric Roessner declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Vitacolonna, M., Belharazem, D., Hohenberger, P. et al. In-vivo quantification of the revascularization of a human acellular dermis seeded with EPCs and MSCs in co-culture with fibroblasts and pericytes in the dorsal chamber model in pre-irradiated tissue. Cell Tissue Bank 18, 27–43 (2017). https://doi.org/10.1007/s10561-016-9606-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10561-016-9606-1