
Abstract
Genetic factors identified from genome-wide association studies have been used to understand causative variants for complex diseases. Studies conducted on large populations of individuals from many geographical regions have provided insights into genetic pathways involved in the causal pathway for atherosclerotic cardiovascular disease. A single genetic trait may ineffectively evaluate the pathway of interest, and it may not account for other complementary genetic pathways that may be activated at various stages of the disease process or evidence-based therapies that alter the molecular and cellular milieu.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Genetic factors identified from genome-wide association studies (GWAS) have been used to understand causative variants for complex diseases. Studies conducted on large populations of individuals from many geographical regions have provided insights into genetic pathways involved in the causal pathway for atherosclerotic cardiovascular disease (CVD) inclusive of lipoprotein metabolism, inflammation and thrombosis. However, traits identified with genome wide significance collectively contribute less than 10 % of the genome wide association risk for coronary artery disease (CAD) [1]. Most often, the proportion of variance explained by the variant is small [2]. Thus, a single genetically-determined trait may ineffectively evaluate the pathway of interest.
Complex pathobiological processes in atherosclerosis involve supernumerary pathways that may have variable effects on the disease state [3]. Genetic studies consider environmental factors as completely independent; however, many genetic factors are dependent on environmental influences [4]. The contexts of shifting environments are not considered in GWAS. Context-driven macroenvironmental factors include lifestyle factors (diet, physical activity, and smoking) and other major risk factors (obesity, diabetes, hypertension) and systemic inflammation [5]. Macroenvironmental factors will influence the cellular microenvironment, which may potentially activate silent inflammatory genes relevant in the late phase of atherosclerosis. The context-dependency of the microenvironmental perturbation may be required for activation of a cotranscription factor that is needed for the regulatory effect of the DNA variant. Thus, genetic variants identified by GWAS have not accounted for environmental context that requires a systems’ genetics approach that identifies disease-driving networks and their genetic regulation.
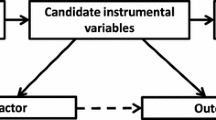
Mendelian randomization is the application of instrumental variables as a tool to infer causality in non-experimental conditions. The instrumental variable is often a GWAS-identified genetic variant or group of variants that associates with both the biomarker of interest and the outcome of interest, but not with other potentially confounding biological pathways [6, 7]. According to the mendelian randomization paradigm, the naturally randomized distribution of genotype occurs at conception and exerts itself throughout the lifetime of the individual. Genetic differences in specific traits are quantified by the presence of one or two specific single nucleotide polymorphisms (SNP). Causality for the trait is implied when its absence is associated with a disease state or clinical event. Most often, the SNP is related to a gene encoding a known intermediary biomarker of interest or target of pharmacological intervention.
Limitations of Mendelian randomization also include confounding by linkage disequilibrium, segregation distortion at the locus of interest, complex genetic architecture, composition of the study population and parent-of-origin effect [7]. Another concern is the gene of interest influences many traits (pleiotropy).
Selection of the instrumental variable in large population based studies and in clinical trials is based on the assumption that the specimen was collected under optimal conditions with high reproducibility and negligible degradation of the selected biomarker during long-term storage. For genetic traits, the biomarker must accurately encompass the pathway of interest and not represent an aggregate measure of the activity of multiple genes that regulate the concentration of different proteins that individually may have pro-atherosclerotic or anti-atherosclerotic activities.
Pharmacogenomics investigates the influence of genetic variants on the response of individuals to a drug. Variants in the target-encoding gene are used to model the potential effects of modulating that target pharmacologically. This approach is intended to define the mechanism-based effects of pharmacological intervention of that target, and distinguish the on-target from off-target effects [8]. Thus pharmacogenomics may be a highly effective tool for selection of personalized and targeted treatment.
In this perspective, we discuss the contributions of the Mendelian randomization paradigm to understanding of biomarkers and targets of therapy. Our examples will review conventional and emerging pathways, therapies, and discuss genetic studies that did not yield the expected outcome. Omission of context-dependent macroenvironment and microenvironmental factors may contribute to overenthusiastic conclusions from GWA traits identified in static environments.
Lipids, Lipoproteins and Lipoprotein Altering Therapies
In this section, genetic targets that regulate Low Density Lipoproteins (LDL), High Density Lipoproteins (HDL) and triglyceride-rich lipoproteins (TGRLs) and pharmacological approaches to these targets will be discussed (Table 1).
LDL Receptor (LDLR)
Rare LDL receptor mutations have been shown to contribute to increased risk of myocardial infarction, while more common variants at nearly 50 loci have been associated with myocardial risk in the general population [9–13]. LDLR mutations have been detected in 2 % of individuals with early-onset MI (≤50 years in males and ≤60 years in females) [13]. More severe LDLR mutations were associated with higher MI risk in the US National Heart Lung, and Blood Institute’s exome sequencing project. Carriers of rare non-synonymous mutations had a 4.2-fold increased risk for MI versus non-MI controls, and carriers of null alleles had a higher 13-fold risk for MI.
LDLR sequence non-coding variants have been shown to be protective against coronary artery disease (CAD) in an Icelandic population [14]. A splice variant in LDLR (rs72658867-A, c.2140 + 5G > A) and an intronic variant (rs17248748-T) are associated with lower non-HDL-C levels and lower coronary artery disease risk. The splice variant alters the LDLR reading frame after amino acid position 713, and increases mRNA expression of LDLR that was unrelated to increased expression in wild type LDLR.
HMG-CoA Reductase and Pharmacological Inhibition
Statins reduce LDL cholesterol levels by inhibiting 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGR), the rate limiting step in cholesterol biosynthesis, and increasing LDL receptor synthesis, the primary receptor for clearance of LDL and other apoB-containing atherogenic lipoproteins. The magnitude of HMG-CoA reduction results in a correlative reduction in cardiovascular events [15]. An analysis of four randomized trials of statin therapy for primary prevention Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) and Anglo-Scandanavian Cardiovascular Outcomes Trial (ASCOT) and secondary prevention Cholesterol And Recurrent Events (CARE) and Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT) that included 48,421 participants who had 3477 events were used to investigate whether a genetic risk score based on 27 genetic variants could ascertain the risk of both incident and recurrent coronary heart disease events [16]. The genetic risk score was selected from traits identified at a genome-wide level in a different analysis [17]. The benefit of statin therapy on incident and recurrent coronary events increased across the low (13 %), intermediate (29 %) and high (48 %) genetic risk categories. The high genetic risk participants had larger absolute risk reduction resulting in a threefold decrease in the number needed to treat to prevent one CHD event in primary prevention trials. In the primary prevention trials, the number needed to treat to prevent one event in 10 years was 25 in those at high genetic risk, 42 in those with intermediate risk and 66 in those at low risk in JUPITER, and 20, 47 and 57 in ASCOT. This approach delineates the expanding role of genetics to provide improved precision for predictors of risk and responses to therapy [18].
Statin therapy is associated with increased risk of type 2 diabetes mellitus [19], that is more pronounced among non-diabetes patients treated with high-intensity compared with low to moderate intensity statin therapy [20]. In the JUPITER trial, treatment with rosuvastatin 20 mg daily increased incident type 2 diabetes by 1.25 [21]. The Mendelian randomization principle was used to investigate the mechanism underlying the glucose-raising effect of statins. Single SNPs in the HMGCR gene, rs17238484 and rs12916, were used to explore the association of incident and prevalent type 2 diabetes [22]. The genetic analysis included 195,444 individuals who participated in 43 studies. The rs172348484G allele was associated with higher plasma insulin concentration (1.62 %, 95 % CI 0.53–2.72; p = 0.04), high plasma glucose concentration (0.23 % 0.02–0.44; p = 0.03) and higher body weight (95 % CI 0.18–0.43; p = 3.15 × 10−6) in 26,236 cases and 164,842 controls from 35 population studies. However, the HMGCR rs1723848-G allele was not associated with increased risk of type 2 diabetes (OR per allele: 1.02, 95 % CI 10.00-1.05; p = 0.09) in 16 studies that included 14,976 cases and 74,395 controls. In contrast, the association between HMGCR rs12916 and incident type 2 diabetes was significant in 16 studies that included 14,976 cases and 74,395 controls). The OR per rs12916-T allele was 1.06 (95 % CI 1.03–1.09; p = 9.58 × 10−5) in 129,170 individuals in randomized trials at a mean 4.2 years follow-up, and increased the odds of new-onset type 2 diabetes (OR 1.12, 95 % CI 1.06–1.18 in all trials). Despite the association of statin therapy and incident type 2 diabetes was reported in the JUPITER trial, the rs17234848 allele was 0.86 (0.70–1.04) rs12916 allele was protective against type 2 diabetes (OR 0.83 [0.70–0.98]) in 17,802 participants (8901 rosuvastatin, 8901 placebo). However, larger studies that included greater numbers of type 2 diabetes patients (WGHS, deCODE) showed estimates that suggested higher risk of type 2 diabetes. Overall, the attributable risk for type 2 diabetes with theses alleles ranged from 2 to 6 %.
PCSK9 and Pharmacological Inhibition
The genetics of proprotein convertase subtilisin kinexin 9 (PCSK9) has identified missense and nonsense loss of function variants that are associated with reduced levels of low density lipoprotein (LDL) cholesterol and lower rates of myocardial infarction [23, 24]. In the pooled cohorts, PCSK9 variants were associated with 35 mg/dL (95 % confidence interval [CI]: 32, 39) lower LDL-C in AAs and 13 mg/dL (95 % CI: 11, 16) lower LDL-C in whites. PCSK9 variants were associated with a pooled OR for CHD of 0.51 (95 % CI: 0.28, 0.92) in AAs and 0.82 (95 % CI: 0.63, 1.06) in whites. These associations support the concept that lifelong lower levels of LDL-C are accompanied by lower risk of CHD. Successful pharmacological approaches to PCSK9 inhibition include the use of monoclonal antibodies directed at circulating levels of PCSK9. In pooled analyses, the completely human monoclonal anti-PCSK9 antibodies (alirocumab, evolocumab) reduces LDL cholesterol levels by 55 to 72 %; and in post hoc analysis of phase II trials, cardiovascular events were reduced by 53 and 54 % respectively [25, 26].
NPC1L1 and Pharmacological Inhibition
Niemann-Pick C1-like1 is expressed in the small intestine and liver, where it functions as a transporter of dietary cholesterol from the intestinal lumen to the enterocytes [27, 28]. Ezetimibe, a pharmacological agent that inhibits the function of NPC1L1, reduced LDL cholesterol levels by 15 to 20 %. The Myocardial Infarction Genetics Consortium Investigators identified 15 naturally occurring inactivating mutations that were associated with an average LDL cholesterol level that was 12 mg/dL lower than in noncarriers [29]. Carriers’ status for an inactivating mutation was associated with a 53 % relative reduction in the risk of coronary heart disease (carrier frequency 0.04 % vs. 0.09 % in controls). Mendelian randomization was used to explore the effects of inhibiting NPC1L1, HMGCR or both on CHD events from 14 prospective and case-control studies that included 108,376 individuals (10.464 CHD events) [30]. NPC1L1 polymorphisms were associated with a 2.4 mg/dL lower LDL-C and 4.8 % lower risk of CHD, while the group with HMRCR polymorphisms had a 2.9 mg/dL lower LDL-C and 5.3 % lower risk of CHD. Among individuals with NPC1L1 and HMGCR polymorphisms had an additive 5.8 mg/dL lower LDL-C and larger 10.8 % lower risk of CHD. This 2 × 2 genetic analysis provides support for NPC1LI as a valid target for pharmacological intervention in combination with statin therapy, which was recently supported by the IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) trial [31]. IMPROVE-IT randomized post acute coronary syndrome patients with LDL cholesterol levels between 50 to 125 mg/dL, to simvastatin 40 mg nightly or simvastatin 40 mg daily and ezetimibe 10 mg daily. After an average of 7 years, treatment with simvastatin and ezetimibe was accompanied by a 6.4 % reduction in major adverse cardiovascular events. Variants in NPC1L1 contribute to the inter-individual variation in LDL cholesterol lowering with ezetimibe; [32] however, the association between the magnitude of LDL cholesterol reduction and cardiovascular events with ezetimibe is uncertain.
CETP and Pharmacological Inhibition
HDL cholesterol has been considered as a biomarker for the efficiency of reverse cholesterol transport, which is the process that results in removal of cholesterol from tissues and disposition in the feces [33]. This concept was based on the lecithin cholesterol acyltransferase pathway promulgated by Glomset nearly 60 years ago [34]. Of the multiple pathways involved in cholesterol transport, the quintessential pathway for removal of cholesterol from lipid-laden macrophage in the arterial wall involves the ABCA1 transporter. However, less than 5 % of the cholesterol transported in HDL particles is derived from the arterial wall. Evaluations of the gene LIPG Asn396Ser that regulates the cholesterol content of high density lipoprotein (HDL) without altering LDL cholesterol, has shown that the cholesterol cargo transported by HDL, e.g., HDL cholesterol concentration, is not involved in the causal pathway for myocardial infarction [35–37]. Consistent with the metabolic interactions with triglyceride-rich lipoproteins, HDL cholesterol risk is related to elevated concentrations of triglyceride-rich lipoproteins, as supported in Mendelian randomization studies [38]. Other HDL protein and lipid components participate in multifarious HDL functions; however, there are no reports from Mendelian randomization studies that have investigated the genetic basis of these components on HDL function and myocardial infarction [37].
Cholesteryl ester transfer protein (CETP) is an important regulator of HDL cholesterol and LDL cholesterol [39]. CETP variants located near or in the CETP gene at 16q13 were associated with higher levels of HDL cholesterol and lower risk of incident myocardial infarction. SNP rs708272 in the CETP gene was associated with a per-allele increase in HDL cholesterol levels of 3.1 mg/dL, and a corresponding 24 % lower risk of future myocardial infarction (age-adjusted HR 0.76, 95 %CI 0.62–0.94). Concordant effects on HDL cholesterol and incident myocardial infarction were also observed at the CETP locus for rs4329913 and rs7202364 [40, 41]. In a meta-analysis, the CETP polymorphism was associated with a decrease in LDL cholesterol levels of 1.2 mg/dL. However, the use of small molecules directed at CETP inhibition has not yet established this target as a viable candidate for CVD prevention [42–44] Despite the failure of clinical trials with certain CETP inhibitors (torcetrapib, dalcetrapib, evacetrapib), it remains to be determined whether other CETP inhibitors do not have hazardous off-target toxicity of torcetrapib or may be more effective than dalcetrapib and evacetrapib agents for lowering LDL cholesterol, triglyceride-rich lipoproteins and increasing HDL cholesterol (anacetrapib, TA-8995) reduce cardiovascular events [45, 46]. However, CETP inhibition with three agents has not been accompanied by a reduction in cardiovascular events suggesting other pathways may interfere with HDL functionality in statin-treated patients [47].
A pharmacogenomics approach has been proposed for the identification of individuals who derive benefit versus harm from CETP inhibition. In the dal-Outcomes trial, polymorphisms in the ADCY9 gene on chromosome 16 were associated with reductions in cardiovascular events [48]. Among patients with genotype AA at rs1967309, there was a 39 % reduction in the composite cardiovascular endpoint with dalcetrapib compared with placebo (hazard ratio, 0.61; 95 % confidence interval, 0.41–0.92). In contrast, patients with genotype GG had a 27 % increase in events with dalcetrapib versus placebo. These data have been replicated in dalPLAQUE with carotid intima-medial thickness. This pharmacogenomic approach to CETP inhibition has been suggested as strategy to investigate whether dalcetrapib may prove an effective target for the prevention of atherosclerotic cardiovascular events among individuals with the certain variants in ADCY9.
ApoCIII and Pharmacological Inhibition
Elevated triglyceride levels have been associated with increased risk of cardiovascular events; however, the causality of triglyceride-rich lipoproteins has been challenging due to the associations with other major cardiovascular risk markers [38]. Two studies have established a central role for apoC3 in triglyceride-rich lipoproteins and CHD risk. In the TG and HDL Working Group of the Exome Sequencing Project, the protein-coding regions (exome) of 18,666 genes were sequenced in 3734 individuals from the United States, and rare mutations in each gene were tested for association with plasma TG [49]. The strongest association was observed for APOC3. Approximately 1 in 150 individuals carried any of 4 protein-altering or splice-site variants of APOC3. Heterozygous carriers of any of these 4 APOC3 mutations had 39 % lower plasma triglyceride levels, 46 % lower circulating plasma APOC3 and 40 % lower risk for CHD. In two general populations studies from Denmark that included data from 75,725 participants, participants with nonfasting TG levels <90 mg/dL had a significantly lower incidence of cardiovascular disease than those with levels ≥350 mg/dL (hazard ratio [HR] for ischemic vascular disease, 0.43; 95 % CI, 0.35 to 0.54; HR for ischemic heart disease, 0.40; 95 % CI, 0.31 to 0.52) [50]. Sequencing of APOC3 in 10,797 participants identified 3 of the 4 loss-of-function mutations from the Exome Sequencing Project. Those heterozygous for loss-of-function mutations in APOC3 had lower triglyceride levels and lower ischemic vascular disease risk that were similar to the first study (44 % nonfasting triglycerides and 41 % for ischemic vascular disease).
In phase 2 studies, an antisense approach to apoCIII, ISIS-APOCIIIRx, was highly effective in lowering apoC3, fasting plasma triglycerides, and non-HDL-C in patients with elevated VLDL-triglyceride or chylomicron-triglyceride due to a variety of conditions, including familial chylomicronemia due to LPL deficiency (LPDL). These data suggest that apoC3 might play a key role in a non-LPL-dependent TGRL metabolic pathway [51, 52].
Inflammatory Pathways and Anti-Inflammatory Therapies
Circulating levels of hepatic-derived inflammatory markers have been associated with increased risk of coronary heart disease in observational epidemiological studies and clinical trials of statins; however, DNA variations associated with C-reactive protein and fibrinogen, have not been linked to CHD in genetic studies [53–55].
Clinical investigations of proximate mediators of inflammatory cascades are challenging due to the large variability in these circulating mediators that are influenced by a multitude of epigenetic changes in gene expression and environmental factors that include the acute phase response. Subsequently, genetic variants of specific inflammatory pathways have been used to assess the causal relationship to CHD. The complexity of mendelian randomization to imply causality in a dynamic environment of atherosclerosis, risk factors and therapies is described for secretory phospholipase A2 (Table 2).
Inflammatory pathways include polymorphisms in lipopolysaccharide-induced inflammatory response; [56] however, genetic associations of the pattern-recognition receptor for LPS (TLR4) are associated with cardiac events [57, 58], but not consistently with atherosclerosis [59, 60]. TLR2 is another recognition receptor for pathogen-associated molecular patterns (PAMPs), and the TLR2 gene Arg753Gln polymorphism was associated with reduced risk of coronary artery disease in Turkish patients [60, 61]. Osteoprotegerin (TNFRSF11B), a secretory glycoprotein member of the tumor necrosis factor receptor (TNFR) superfamily, inhibits differentiation of macrophages into osteoclasts tumor necrosis factor soluble factor 11B [62]. In peripheral arterial disease patients with diabetes, three variant genotypes of the OPG gene (rs3134069, rs2073617, rs2073618) were independently associated with increased risk of peripheral arterial occlusive disease, but these three traits act synergistically with different levels of risk. This study suggests that the susceptibility profile results from functional interactions between a number of genes.
Interleukin-6 Receptor Pathways
The interleukin-6 (IL6) signaling pathway involves soluble IL-6 activation of hepatic, leukocyte and endothelial membrane-bound receptors (IL6R) that result in activation of downstream inflammatory cascades that increase production of hepatically-synthesized C-reactive protein, fibrinogen, and other acute phase reactants [63, 64]. The Asp358Ala variant, rs2228145, in IL6R impairs IL6R signaling by reducing membrane bound IL6R levels, and not by changing production of IL6R or IL-6 [65, 66].
Two Mendelian randomization meta-analyses have investigated IL-6R and CHD [67, 68]. In a meta-analysis of human genetic and biomarker data from 46 genetic epidemiological studies involving 204,930 participants, carriers of 358Ala had higher concentrations of soluble IL6R and interleukin 6 (both p < 10−13) and dose-dependent lower concentrations of the downstream inflammatory markers CRP and fibrinogen, compared to non-carriers [67]. The risk of CHD was lower in carriers of 358Ala compared with non-carriers. Carriers had a 3.4 % (1.8–5.0) per allele lower risk of CHD. In another mendelian randomization study that included 133,449 participants from 40 studies, the IRLR SNP rs7529229 marking a IL6 variant rs8192284 (p.Asp358Ala) was associated with an increased circulating concentration of IL-6 (log increase in concentration 9.45 % per allele) and reduced concentrations of CRP (8.35 % [95 % CI: 8.34–10.57] per allele) and fibrinogen (0.85 % [95 % CI: 0.60–1.10] per allele) [68]. The IL6R rs7529229 SNP was associated with reduced odds of CHD events (0.95: 95 % CI: 0.93, p = 1.53 × 10−5).
In an example of pharmacogenomics, the genetic findings of IL6R polymorphism rs7529229 were compared with the aggregated data from rheumatoid arthritis patients who were enrolled in six short-term randomized or controlled trials (12–52 week duration) with the IL-6R monoclonal antibody blocker tocilizumab [68]. Treatment with toclizumab (8 kg/mg dose) reduced IL-6, IL-6R, CRP and fibrinogen, but increased LDL cholesterol and HDL cholesterol. The IL6R rs7529229 SNP showed no association with LDL cholesterol and HDL cholesterol. The effects of tolizumab may be mechanism based due to reduced inflammation and alleviation of acute phase lipoprotein changes or result from differences in classic signaling through membrane IL6R versus trans-signaling via the soluble receptor. Tolizumbab inhibits classical and trans pathways, whereas the functional polymorphism tagged by rs7529229 (rs8192284) increases soluble ILR6R only through proteolytic cleavage of the membrane-bound receptor [64, 69, 70].
Secretory Phospholipase A2
Secretory phospholipase A2 encompasses a family of calcium-dependent phospholipase that include members with pro-atherogenic (sPLA2-IIA, sPLA2-III and sPLA2-V) and anti-atherogenic properties (sPLA2-X) [70–74]. The sPLA2 isoenzymes, GII, V ad X, participate in intracellular arachidonic acid release and as a consequence influence intracellular inflammatory signaling and production of bioactive lipid mediators by different pathways [71, 73]. Each isoform functions both as an enzyme and ligand for receptors (both soluble and membrane-bound) that control and transduce their biological effects or modulate their enzymatic action [70–72]. Among the sPLA2 binding proteins, the M-type receptor (PLA2R1) is the best-characterized sPLA2 receptor [75, 76]. PLA2R1 is a type I membrane glycoprotein of 180 kDa that belongs to the superfamily of C-type lectins. Currently, the biological functions of PLA2R1, its endogenous ligands, other than mammalian sPLA2 isoenzymes, and associated signaling pathways remain largely unknown. In an experimental model, lack of PLA2R1 increased the risk of cardiac rupture after myocardial infarction [77].
During an acute coronary syndrome, the plasma concentration of sPLA2-IIA rises, and this biomarker has been shown useful to discriminate between suspected ACS patients who develop a myocardial infarction versus those patients who do not [71–73]. The localization pattern of sPLA2-IIA in infarcted myocardium and its temporal course in plasma, in relation to those of CRP, are in line with a supposed pro-inflammatory role during acute myocardial infarction for sPLA2-IIA, as a source of lysophospholipids that serve as ligands for CRP [78]. This example with sPLA2-IIA underscores the importance of context dependency [5]. (Bjorkman).
Varespladib methyl is a pan-sPLA2 inhibitor that has near equal efficacy in lowering production of sPLA2-IIA, sPLA2-V and sPLA2-X [79]. Thus, this compound reduces the concentration of both proatherosclerotic and anti-atherosclerotic sPLA2 isoforms. In a biomarker analysis of ACS patients, varespladib methyl reduced LDL cholesterol and high-sensitivity C-reactive protein; [80] two biomarkers that were consistent predictors of benefit with multiple pharmacological therapies [81]. In a subgroup analysis of the Fewer Recurrent Acute coronary events with Near-term Cardiovascular Inflammation Suppression (FRANCIS) trial, the anti-inflammatory effect of varespladib methyl was confined to patients with diabetes [82], thereby providing another example of context-dependent macroevironmental influences and most certainly microenvironmental influences. VISTA-16 (Vascular Inflammation Suppression to Treat Acute coronary syndromes for 16 weeks) investigated the short-term efficacy of varespladib methyl therapy (500 mg daily) on cardiovascular morbidity and mortality when added to standard of care and atorvastatin [83, 84]. The trial was terminated due to possible harm as demonstrated by the 1.66 hazard ratio for myocardial infarction.
The association between secretory phospholipase A2 isoforms as potential therapeutic targets for cardiovascular disease prevention was subsequently evaluated in Mendelian randomization studies. Various SNPs of PLA2GA (rs1774131, rs11573156, rs3753827, rs2236771; rs876018; rs3767221) influence sPLA2-IIA concentration in patients with stable CHD; however, only linkage to rs11573156 was associated with sPLA2-IIA level in multivariate analysis [85]. None of the PLA2GA SNPs were associated with total sPLA2 activity. Due to the lack of available assays to directly quantify sPLA2-V levels, the sPLA2-V Mendelian randomization used expression data to select the SNP most strongly associated with PLA2G5 gene expression [86]. The association of the genetic variant was investigated in a pooled analysis of 27,230 CHD events and 70,500 controls. There was no evidence to support the role of sPLA2-V in CHD. A smaller genetic analysis of sPLA2-X suggested no evidence of association of the sPLA2-X isoform with risk of CHD [87].
These Mendelian randomization studies have questioned whether sPLA2 isoforms have a causal effect on CHD. Several assumptions related to the biology of sPLA2 isoforms and pharmacological effects of varespladib methyl require further consideration regarding interpretation of Mendelian randomization studies as illustrated in (1) absolute values for sPLA2-IIA levels and activity were not reported [88], which is important due to variable results with different analytical methods; (2) wide differences in baseline sPLA2-IIA levels in different cohorts, and marked differences in sPLA2 levels that results from the acute phase reaction in ACS patients versus stable CHD patients; (3) use of total sPLA2 activity as a surrogate for sPLA2-IIA activity does not account for the anti-atherogenic and pro-atherogenic contributions of other sPLA2 isoforms such as groups III, V and X sPLA2; (4) marginally significant correlation between PLA2G2A rs11573156 variant and sPLA2 activity despite the higher correlations between sPLA2-IIA mass and sPLA2 activity; (5) biomarker effects were reported FRANCIS where the protocol mandated that all patients have their statin therapy changed to atorvastatin 80 mg daily regardless of their prior statin regimen. However, the use of Mendelian randomization studies to deduce pharmacological effects does not account for the properties of the specific inhibitor. Specifically, varespladib methyl is: (1) hydrophilic and may not penetrate into vascular tissues with sufficient potency to reduce intracellular effects versus the consistent effects on plasma biomarkers; (2) a pan sPLA2 inhibitor with similar efficacy in lowering groups IIA, and X sPLA2 with somehow lower potency against group V; thus, varespladib methyl inhibits sPLA2-X, an atheroprotective isoform; (3) genetic studies do not account for intracellular effects of sPLA2 inhibition on eicosanoid production that may have net prothrombotic or antithrombotic effects; and (4) VISTA-16 reported an increase in myocardial infarctions in varespladib methyl treated patients, and this harmful effect of loss-of-function variants was not observed in the mendelian randomization studies.
Lipoprotein Associated Phospholipase A2 and Pharmacological Inhibition
Lipoprotein associated phospholipase A2 (Lp-PLA2) provides an example where genetic studies were inconsistent and limited to certain regions of the world. Although meta-analyses including more than 13,000 subjects from 5 population-based studies variants at two loci (PLA2G7, CETP) were associated with Lp-PLA2 mass and 4 out of 6 top SNP were associated with Lp-PLA2 activity (ZNF259, APOC1, CELSR2 SCARB1), the two SNPs in the PLA2G7 gene most significantly associated with mass, did not show a significant association with CHD/CAD in the large CARDIoGRAM consortium [89]. In the two pivotal trials, STABILITY (Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy) [90] and SOLID (Stabilization of Plaque Using Darapladib-Thrombolysis in Myocardial Infarction 52) [91], selective inhibition of Lp-PLA2 inhibition with darapladib had no effect on CVD outcomes. High-intensity statin therapy may have reduced the chances of efficacy through reduction in substrate (apoB-containing lipoproteins, oxidatively modified LDL particles), indirect effects on Lp-PLA2 mass and activity, and anti-inflammatory properties independent of Lp-PLA2 [92].
Conclusions
Mendelian randomization studies have identified traits involved in lipoprotein metabolism and inflammation that serve as targets for therapeutic intervention. The LDL receptor regulating genetic traits, HMGCR and NPC1L1, are examples where Mendelian randomization and clinical trials are congruent. Although clinical trials with PCSK9 inhibitors are ongoing, two post hoc analyses provide support for targeting proteins that regulate LDL receptor activity. In contrast, CETP variants associated with lower LDL-C and higher HDL-C levels have not been proven effective in several clinical trials with CETP inhibitors. Pharmacogenomic analyses of adverse events has produced inconsistent results as illustrated in the GWAS study of HMGR and incident type 2 diabetes where x trait was positively associated with type 2 diabetes in many trials but negatively associated in JUPITER.
Identification of inflammatory targets may be more complex as pro-inflammatory and anti-inflammatory pathways are redundant, and inflammatory responses involve cellular receptors and intracellular signaling pathways. Mendelian randomization with secretory phospholipase A2 variants provides an example where genetic analyses were congruent with the pro-atherosclerotic properties of sPLA2-IIA and sPLA2-V, but not with the anti-atherosclerotic properties of sPLA2-X. Clinical trials of phospholipase A2 inhibitors provide an example for the need of system genetics to study inflammatory inhibitors at various stages of the disease process and accounts for the effect of background secondary preventive therapy, such as high-intensity statin therapy on the substrate, enzyme activity and effectors that is the target of the selective inhibitor.
Selection of major polymorphisms associated with certain lipoprotein and inflammatory biomarkers cannot account for the multitude of variables that regulate gene expression or post-translational modification of gene product. The use of pharmacogenomics for selection of inhibitors that target a causal pathway may minimize risk of pharmaceutical development, but other considerations may impact the success of a therapeutic target including concomitant therapy that reduces substrate availability and enzyme activity; vascular effects that have a direct or indirect impact on the disease process, tissue penetration, and off-target toxicity of the selective inhibitor.
References
DeLoukas P, Kanoni S, Willenborg C, for the CARDioGRAMplusD4D consortium, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33.
Roberts R. Genetics of coronary artery disease. Circ Res. 2014;114:1890–903.
Rosenson RS. Future role of selective phospholipase A2 inhibitors in the prevention of atherosclerotic cardiovascular disease. Cardiovasc Drugs Ther. 2009;23:93–101.
Schradt EE, Bjorkegren JL. NEW: network-enabled wisdom in biology, medicine, and health care. Sci Transl Med. 2012;4:115rv1.
Bjorkegren JLM, Kovacic JC, Dudley JT, Schadt EE. Genome-wide significant loci: how important are they? J Am Coll Cardiol. 2015;65:830–45.
Greenland S. An introduction to instrumental variables for epidemiologists. Int J Epidemiol. 2000;29:722–9.
Ridker PM, Paynter NP, Danik JS, Glynn RJ. Interpretation of Mendelian randomization studies and the search for causal pathways in atherothrombosis: the need for caution. Metab Syndr Relat Disord. 2010;8:465–9.
Sofat R, Hingorani AD, Smeeth L, et al. Separating the mechanism-based and off-target actions of cholesteryl ester transfer protein inhibitors with CETP gene polymorphisms. Circulation. 2010;121:52–62.
McPherson R, Pertsemlidis A, Kavaslar N, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316(5830):1488–91.
Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357(5):443–53.
Helgadottir A, Thorleifsson G, Manolescu A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316(5830):1491–3.
Kathiresan S, Voight BF, Purcell S, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41(3):334–41.
Do R, Stitziel NO, Won HH, Jørgensen AB, Duga S, Angelica Merlini P. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518:102–6.
Gretarsdottir S, Helgason H, Helgadottir A, Sigurdsson A, Thorleifsson G, Magnusdottir A, et al. A splice region variant in LDLR lowers Non-high density lipoprotein cholesterol and protects against coronary artery disease. PLoS Genet. 2015;11(9), e1005379. doi:10.1371/journal.pgen.1005379.
Cholesterol Treatment Trialists’ (CTT) Collaboration. The effects of lowering LDL cholesterol with statin therapy in people at low risk for vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–90.
Mega JL, Stitzel NO, Smith JG, et al. Genetic risk, coronary heart disese evetns, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet. 2015;385:2264–71.
Schunkert H, Konig IR, Kathiresan S, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–8.
Schunkert H, Samani N. Statin treatment: can genetics sharpen the focus? Lancet. 2015;385:2227–9.
Sattar N, Preiss D, Murray HK, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomized statin trials. Lancet. 2010;375:735–42.
Preiss D, Seshasai SRK, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA. 2011;305:2556–64.
Ridker PM, Pradhan A, MacFadyen JG, Libby P, Glynn RJ. Cardiovascular benefits and diabetes risks for statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet. 2012;380:565–71.
Swerdlow D, Preiss D, Kuchenbaecker KB, et al. HMG-coenzyme A reductase inhibition, type 2 diabetes and bodyweight: evidence from genetic analysis and randomized trials. Lancet. 2015;385:351–61.
Cohen JC, Boerwinkle E, Mosley Jr TH, et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72.
Kent ST, Rosenson RS, Avery CL, et al. PCSK9 loss-of-function variants, low-density lipoprotein cholesterol, and risk of coronary heart disease and stroke: data from the REGARDS Study and CHARGE Consortium. Circulation 2015;132:A9793.
Robinson JG, Farnier M, Krempf M, Bereron J, Luc G, Averna M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–99.
Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, for the Open-Label Study of Long-Term Evaluation against LDL-Cholesterol (OSLER) investigators, et al. Efficacy and safety of evolocumbab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1500–9.
Davis Jr HR, Zhu LJ, Hoos LM, et al. Niemann-Pick C1 Like 1 (NPC1L1) is the intestinal phytosterol and cholesterol transporter and a key modulator of whole-body cholesterol homeostasis. J Biol Chem. 2004;279:33586–92.
Altmann SW, Davis Jr HR, Zhu LJ, et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol aborption. Science. 2004;303:1201–4.
Stitziel NO, Won H-H, Morrison AC, The Myocardial Infarction Genetics Consortium Investigators. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med. 2014;371:2072–82.
Ference BA, Majeed F, Penmetcha R, Flack JM, Brook RD. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both. A 2 × 2 factorial mendelian randomization study. J Am Coll Cardiol. 2015;65:1552–61.
Cannon CP, Blazing MA, Giugliano RP, IMPROVE-IT Investigators, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–97.
Hegele RA, Guy J, Ban MR, Wang J. NPC1L1 haployte is associated with inter-individual variation in plasma low-density lipoprotein response to ezetimibe. Lipids Health Dis. 2005;4:16.
Rosenson RS, Brewer Jr HB, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–19.
Glomset JA. The plasma lecithin: cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–67.
Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80.
Rosenson RS, Brewer Jr HB, Ansell B, et al. Translation of high-density lipoprotein function into clinical practice: current prospects and future challenges. Circulation. 2013;128:1256–67.
Rosenson RS, Brewer HB Jr., Ansell BJ, et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol 2016;13:48–60.
Rosenson RS, Davidson MH, Hirsh BJ, Kathiresan S, Gaudet D. Genetics and causality of triglyceride-rich lipoproteins in atherosclerotic cardiovascular disease. J Am Coll Cardiol. 2014;64:2525–40.
Barter PJ, Rye K-A. Cholesteryl ester transfer proten inhibition as a strategy to reduce cardiovascular risk. J Lipid Res. 2012;53:1755–66.
Thompson A, Di Angelantonio E, Sarwar N, et al. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA. 2008;299:2777–88.
Ridker PM, Pare G, Parker AN, Zee RYL, Miletich JP, Chasman DI. Polymorphism in the CETP gene region, HDL cholesterol, and risk of future myocardial infarction: genomewide analysis among 18,245 initially healthy women from the Women’s Genome Health Study. Circ Cardiovasc Genet. 2009;2:26–33.
Barter PJ, Caulfield M, Eriksson M, ILLUMINATE Investigators, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22.
Schwartz GG, Olsson AG, Abt M, dal-OUTCOMES Investigators, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–99.
Lilly press release October 12, 2015. Visit:http://www.prnewswire.com/news-releases/lilly-to-discontinue-development-of-evacetrapib-for-high-risk-atherosclerotic-cardiovascular-disease-300157604.html.
Cannon CP, Shah S, Dansky HM, Determining the Efficacy and Tolerability Investigators, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–15.
Hovingh GK, Kastelein JJ, van Deventer SJ, et al. Cholesterol ester transfer protein inhibition by TA-8995 in patients with mild dyslipidaemia (TULIP): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet. 2015;386:452–60.
Rosenson RS, Brewer Jr HB. New challenges for HDL-modifying therapies as a strategy to lower cardiovascular disease events in statin-treated patients. Cardiovasc Drugs Ther. 2015;29:1–3.
Tardif JC, Rhéaume E, Lemieux Perreault LP, et al. Pharmacogenomic determinants of the cardiovascular effects of dalcetrapib. Circ Cardiovasc Genet. 2015;8:372–82.
The TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–3.
Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, et al. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41.
Gaudet D, Alexander VJ, Baker BF, et al. Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N Engl J Med. 2015;373:438–47.
Gaudet D, Brisson D, Tremblay K, Alexander VJ, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med. 2014;371:2200–6.
C-Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC). Association between C reactive protein and coronary heart disease: menedelian randomization analysis based on individual participant data. BMJ. 2011;342.
Elliott P, Chambers JC, Zhang W, et al. Genetic loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302:37–48.
Keavney B, Danesh J, Parish S, et al. Fibrinogen and coronary heart disease: test of causality by “Mendelian randomization”. Int J Epidemiol. 2006;35:935–43.
Li L, He M, Zhou L, Miao X, et al. A solute carrier family 22 member 3 variant rs3088442 G → A associated with coronary heart disease inhibits lipopolysaccharide-induced inflammatory response. J Biol Chem. 2015;290:5328–40.
Amerziane N, Beillat T, Verpillat P, et al. Association of the Toll-like recpeotor 4 gene Asp299Gly polymorphism with acute coronary events. Arterioscler Thromb Vasc Biol. 2003;23:e61–4.
Ballistreri CR, Candore G, Colonna-Romano G, et al. Roll of Toll-like receptor 4 in acute myocardial infarction and longevity. JAMA. 2004;292:2339–40.
Kolek MJ, Carlquist JF, Muhlestein JB, et al. Toll-like receptor 4 gene Asp299Gly polymorphism is associated with reductions in vascular inflammation, angiographic coronary artery disease, and clinical diabetes. Am Heart J. 2004;148:1034–40.
Guven M, Ismailoglu Z, Batar B, et al. The effect of genetic polymorphisms of TLR2 and TLR4 in Turkish patients with coronary artery disease. Gene. 2015;558:99–102.
Alarcon G-V, Martinez J A, Villarreal-Molina T, et al. Interleukin-17A gene haplotypes are associated with risk of premature coronary artery disease in Mexican patients from the Genetics of Atherosclerotic Disease (GEA) study. PLoS One 2015.
Biscetti F, Porreca CF, Bertucci F, et al. TNFRSF11B gene polymorphisms increased risk of peripheral arterial occlusive disease and critical limb ischemia in patient with type 2 diabetes. Acta Diabetol. 2014;51:1025–32.
Rosenson RS. Myocardial injury, the acute phase response and lipoprotein metabolism. J Am Coll Cardiol. 1993;22:933–40.
Jones SA, Horiuchi S, Topley N, Yamamoto N, Fuller GM. The soluble interleukin 6 receptor: mechanisms of production and implications of disease. FASEB J. 2001;15:43–58.
Reich D, Patterson N, Ramesh V, the Health, the Health, Aging and Body Composition (Health ABC) Study, et al. Admixture mapping of an allele affecting interleukin 6 soluble receptor and interleukin 6 levels. Am J Hum Genet. 2007;80:716–26.
Stephens OW, Zhang Q, Qu P, et al. An intermediate-risk multiple myeloma subgroup is defined by sIL6r levels synergistically increase with incidence of SNP rs2228145 and 1q21 amplification. Blood. 2012;119:503–12.
IL6R Genetic Consortium and Emerging Risk Factors Collaboration. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet. 2012;379:1205–13.
The Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisaton analysis. Lancet. 2012;379:1214–24.
Melton L. Coobs, A, Actemra poised to launch IL-6 inhibitors. Nat Biotechnol. 2008;26:957–9.
Burke JE, Dennis EA. Phospholipase A2 biochemistry. Cardiovasc Drugs Ther. 2009;23:49–59.
Rosenson RS, Gelb MH. Secretory phospholipase A2: a multifaceted family of proatherogenic enzymes. Curr Cardiol Rep. 2009;11:445–51.
Rosenson RS. Phospholipase A2 inhibition and atherosclerotic vascular disease: Prospects for targeting secretory and lipoprotein associated phospholipase A2 enzymes. Curr Opin Lipidol. 2010;21:473–80.
Rosenson RS, Hurt-Camejo E. Phospholipase A2 enzymes and the risk of atherosclerosis. Eur Heart J. 2012;33:2899–909.
Ait-Oufella H, Herbin O, Lahoute C, et al. Group X secreted phospholipase A2 limits the development of atherosclerosis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol. 2013;33:466–73.
Ishizaki J, Hanasaki K, Higashimo K, et al. Molecular cloning of pancreatic group I phospholipase A2 receptor. J Biol Chem. 1994;269:5897–904.
Lambeau G, Ancian P, Barhanin J, Lazdunski M. Cloning and expression of a membrane receptor for secretory phospholipases A2. J Biol Chem. 1994;269:1575–8.
Mishina H, Watanabe K, Tamaru S, et al. Lack of phospholipase A2 receptor increases susceptibility to cardiac rupture after myocardial infarction. Circ Res. 2014;114:493–504.
Nijmer R, Lagrand W, Baidoshvili A, et al. Secretory type II phospholipase A2 binds to ischemic myocardium during myocardial infarction in humans. Cardiovasc Res. 2002;53:138–46.
Rosenson RS, Fraser H, Hislop H, Trias J. Varespladib methyl in cardiovascular disease. Exp Opin Invest Drugs. 2010;10:1245–55.
Rosenson RS, Elliot M, Stasiv Y, Hislop C. Effects of varespladib on biomarkers and major cardiovascular events in acute coronary syndrome patients. J Am Coll Cardiol. 2010;56:1079–88.
Rifai N, Ridker PM. High-sensitivity C-reactive protein: a novel and promising marker of coronary heart disease. Clin Chem. 2011;47:403–11.
Rosenson RS, Fraser H, Goulder MA, Hislop C. Anti-inflammatory effects of verespladib methyl in diabetic patients with acute coronary syndrome. Cardiovasc Drugs Ther. 2011;25:539–44.
Nicholls SJ, Cavender MA, Kastelein JJ, et al. Varespladib and cardiovascular events in patients with an acute coronary syndrome: the VISTA-16 randomized clinical trial. Cardiovasc Drugs Ther. 2012;26:71–5.
Nicholls SJ, Kastelein JJ, Schwartz GG, for the VISTA-16 investigators, et al. Varespladib and cardiovascular events in patients with an acute coronary syndrome: the VISTA-16 randomized clinical trial. JAMA. 2014;311:252–62.
Holmes MV, Simon T, Exeter HJ, et al. Secretory phospholipase A(2)-IIA and cardiovascular disease: a mendelian randomization study. J Am Coll Cardiol. 2013;9(62):1966–76.
Holmes MV, Exeter HJ, Folkersen L, et al. Novel genetic approach to investigate the role of plasma secretory phospholipase A2 (sPLA2)-V isoenzyme in coronary heart disease: modified Mendelian randomization analysis using PLA2G5 expression levels. Circ Cardiovasc Genet. 2014;7:144–50.
Guardiola M, Exeter HJ, Perret C, et al. PLA2G10, gene variants, sPLA2 activity, and coronary heart disease risk. Circ Cardiovasc Genet. 2015;8:356–62.
Rosenson RS, Hurt-Camejo E. Letter to the Editor: limits of mendelian randomization analyses in selection of secretory phospholipase A2-IIA as a valid therapeutic target for cardiovascular disease prevention. J Am Coll Cardiol. 2014;63:942–3.
Grallert H, Dupuis J, Bis JC, et al. Eight genetic loci associated with variation in lipoprotein-associated phospholipase A2 mass and activity and coronary heart disease: meta-analysis of genome-wide association studies from five community-based studies. Eur Heart J. 2012;33:238–51.
Thompson A, Gao P, Orfei L, et al. Lp-PLA(2) studies collaboration. Lipoprotein-associated phospholipase a(2) and risk of coronary disease, stroke, and mortality. Lancet. 2010;375:1536–44.
O’Donoghue ML, Braunwald E, White HD, SOLID-TIMI 52 Investigators, et al. Effect of darapladib on major coronary events after an acute coronary syndrome: the SOLID-TIMI 52 randomized clinical trial. JAMA. 2014;312:1006–15.
Rosenson RS, Stafforini DM. Modulation of oxidative stress, inflammation, and atherosclerosis by lipoprotein-associated phospholipase A2. J Lipid Res. 2012;53:1767–82.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rosenson, R.S., Koenig, W. Mendelian Randomization Analyses for Selection of Therapeutic Targets for Cardiovascular Disease Prevention: a Note of Circumspection. Cardiovasc Drugs Ther 30, 65–74 (2016). https://doi.org/10.1007/s10557-016-6642-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-016-6642-9