
Abstract
Purpose of Review
Mendelian randomization (MR) is a technique that uses natural genetic variation to assess the potential causal role of a modifiable risk factor on cardiovascular disease. Advances have led to the identification of single nucleotide polymorphisms linked with risk factors that act as naturally randomized instruments to investigate the risk factor-disease relationship.
Recent Findings
There are several pros and cons when using MR. It can address many limitations of observational study design including confounding, reverse causation, and demonstration of causality when a randomized controlled trial is not practical or feasible. However, several limitations do exist and include pleiotropy (multiple downstream effects of a single genetic variant), linkage disequilibrium (non-random association of genetic variation), and imprecise estimates of causal effects.
Summary
MR is an important tool in cardiovascular research and has been applied to assess cardiovascular risk factors including obesity and atrial fibrillation. While these studies provide insight into disease causation, understanding the strengths and limitations of the technique is important for appropriate interpretation of results.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Prior to the introduction of randomized control trials (RCTs), physicians relied on personal experience, case reports, case series, and epidemiological associations to guide clinical practice. With the introduction of the RCT, researchers established a “gold standard” method for addressing important clinical questions that have changed the practice of medicine. In addition to limiting various biases associated with observational study design, RCTs can go beyond epidemiological associations to establish a causal relationship between a risk factor and a disease. While RCTs have improved our understanding of risk factors and treatments for cardiovascular disease (CVD), this method has several limitations. RCTs can be expensive and time consuming to conduct. They also require rigorous trial design to accurately interpret the results. From an ethical standpoint, clinical equipoise must be established in order to carry out a RCT. By this principle, there must be uncertainty in each arm as to which exposure or treatment will benefit patients. This requirement can make RCTs infeasible to conduct or can limit patient recruitment based upon patient or physician perceptions of benefit and risk. For example, testing the causal effect of a risk factor such as smoking or excessive alcohol use on CVD through a RCT would not be possible as it is not ethical to randomize patients to a smoking or alcohol use arm. Another important limitation to RCTs is that in many cases, there are no available therapeutic agents that can selectively modify the risk factor of interest. This limits the ability of RCTs to assess whether a risk factor is truly causal.
The limitations of RCTs to establish causality have led to the development of alternative techniques to investigate the relationship between a modifiable risk factor and a disease outcome. Mendelian randomization is one such technique that has gained increasing interest. Use of Mendelian randomization was initially utilized by Martin Katan to understand the relationship between cholesterol levels and cancer morbidity/mortality [1]. Since an individual’s cholesterol levels are inherently confounded by comorbidities that accompany cancer, Katan proposed to use genetic variation in the apolipoprotein E (ApoE) gene that correlates with low-density lipoprotein cholesterol (LDL-C) and total cholesterol levels, rather than the cholesterol levels themselves. He hypothesized that, if low cholesterol was indeed causally related to development of cancer, individuals with genetic variation linked to low LDL-C levels would have the highest incidence of cancer. Since Katan’s initial description, Mendelian randomization studies have grown exponentially (Fig. 1), in part, by advances in DNA sequencing and genome-wide association studies (GWAS) that have led to the identification of many of single nucleotide polymorphisms (SNPs) that are linked to risk factors. These variants can be used to answer questions which are not feasible to address by a RCT. It can also limit observational bias and get closer to establishing causality. In this review, we will discuss Mendelian randomization as an epidemiologic tool to evaluate emerging cardiovascular risk factors and describe its strengths and weaknesses. We will also summarize the current literature using Mendelian randomization in cardiovascular disease research and address future directions and applications of this methodology.

Mendelian randomization studies in PubMed. Graphical representation of the frequency of Mendelian randomization studies per year published in PubMed (accessed July 26, 2017)
Understanding Mendelian Randomization
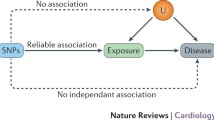
Mendelian randomization uses the random distribution of naturally occurring SNPs that are strongly linked to a risk factor as the instrument for randomization (Fig. 2). This method is founded on the principle that an individual’s genotype is determined randomly at conception from his/her parental genotypes and that genetic variants governing variation in one trait are (often) inherited independently of those influencing another trait. Therefore, genotype will in most cases be independent of behavioral, dietary, and other factors that could confound the association between risk factor and outcome [2, 3]. Furthermore, unlike most observational studies, those with and without the specific genetic variant are expected to be balanced with regard to both measured and unmeasured confounders. Take for example the use of Mendelian randomization to understand risk factors for CVD through lipoprotein (a) (Lp(a)). Observational studies of Lp(a) have shown inconsistent associations between Lp(a) and CVD [4,5,6]. Lp(a) concentrations can vary 1000-fold with a skewed distribution in most populations and may be influenced by several confounding factors, such as age, sex, lifestyle factors, fasting state, and inflammation. Since the variation in Lp(a) levels is partially controlled by polymorphisms in the LPA gene locus, Mendelian randomization was employed in the Copenhagen City Heart Study using variants in the LPA gene characterized by the number of KIV-2 repeats. They found a modestly increased risk for CVD in carriers of genotypes associated with increased Lp(a) levels, supporting a causal role of elevated Lp(a) in the development of CVD [7].

Using Mendelian randomization to understand the relationship between risk factors and disease. This image depicts the relationship between a genetic variant (single nucleotide polymorphism or SNP), modifiable risk factor (exposure), and disease, accounting for confounders. Mendelian randomization relies on the assumption that genetic variants explain variation in the exposure but do not affect the disease outcome except potentially through the exposure, making them valid instrumental variables. Mendelian randomization is not (presumably) affected by confounders that can be associated with both the exposure and outcome, thereby providing an advantage over traditional observational study design methodology
An important contribution of Mendelian randomization is its ability to circumvent the problem of confounding and assess the causal relationship between an exposure and a disease. However, several assumptions must be met to determine causality. First, causal inferences require a true association between a genetic variant and an exposure, such that a genetic variant can serve as a reliable proxy for the exposure. This assumption is mostly likely to be true when the variant occurs in a gene that is directly related to the exposure such as the sickle cell variant in the HBB gene causing hemoglobin S production and the classic “sickling” changes in the red blood cell. Second, causal inferences require that the relationship between a genetic variant and an outcome is mediated through the exposure. The genetic variant should not affect the outcome directly or through alternate pathways other than the effect mediated by the exposure. For example, for Lp(a) above, the LPA gene would not be an appropriate choice for randomization if it also influenced blood pressure or glucose levels. Similarly, the genetic variant should not be associated with confounders of the outcome. If these assumptions are met and the limitations of Mendelian randomization are considered, evidence of a causal relationship between an exposure and an outcome is made.
Strengths of Implementing Mendelian Randomization
While observational studies have been a useful tool to assess associations between risk factors and disease, there are significant limitations to their use. Observational studies are prone to confounding (a second exposure is related to both the first exposure and the outcome making it seem as if the first exposure and outcome were directly linked), reverse causation (the outcome may actually cause the putative exposure), or selection bias (the exposure is only associated with the outcome in a segment of the population due to alternative characteristics). Mendelian randomization is a way to overcome these limitations since the central principle of this technique is that genetic variants randomly segregate during meiosis. As a result, genetic variants are randomly distributed within a population limiting the presence of confounding factors when participants are stratified by genotype rather than the level of risk factor.
Additionally, Mendelian randomization eliminates the possibility of reverse causation influencing the relationship between a risk factor and an outcome. In observation studies, the direction of the relationship between a risk factor and an outcome cannot be determined. Using Mendelian randomization, genetic variants are inherited prior to the outcome, and genotype stays constant throughout life. As a result, the risk factor or biomarker of interest clearly precedes the development of disease. By this principle, Mendelian randomization can also assess the cumulative lifetime effect of a risk factor such as lifetime exposure LDL-C levels on CVD risk. This addresses the limitations of evaluating the effect of longitudinal exposures on an outcome in observational studies, which have limited exposure/follow-up time.
Mendelian randomization has other advantages over RCTs as well. Although traditional RCTs are the gold standard for assessing the relationship between an exposure and an outcome, they can be difficult to implement or may not be feasible. Publicly available data from GWAS studies can be used to determine the effect of variants associated with a risk factor on outcomes. These variants can then be tested in other populations or in publicly available datasets to determine the causal relationship between a risk factor and a disease, which is often much less costly than designing a RCT.
Limitations of Implementing Mendelian Randomization
While Mendelian randomization can have significant advantages when implemented properly, this study design has certain challenges which can hamper the interpretation of the results. These limitations are important to be aware of then assessing the causal relationship between an exposure and an outcome. One of the most significant limitations is pleiotropy. Pleiotropy is the production of multiple effects by a single variant, all of which can influence the outcome. Vertical pleiotropy occurs when the exposure associates with multiple biomarkers on the same pathway. Horizontal pleiotropy occurs when the exposure associates with multiple biomarkers on different pathways. An example of this type of pleiotropy occurred with the study of a variant in the APOE gene. This variant was found to be associated with C-reactive protein (CRP) levels and risk of CVD, thereby leading the investigators to the erroneous conclusion that CRP was causal for CVD [8••]. However, true causal factor was LDL-C, which is also influenced by the APOE genotype and causally related to CVD. Pleiotropy is least likely to occur when the variant occurs in the gene coding for the biomarker associated with the variant. Pleiotropy becomes more likely if the relationship between the variant and the outcome is complex or indirect. Absence of pleiotropy is an assumption of Mendelian randomization and, if a SNP has pleiotropic effects, it is not a valid instrument and cannot be used. However, an alternative SNP could still be chosen as a proxy for the exposure allowing a Mendelian randomization approach.
A second limitation of Mendelian randomization is linkage disequilibrium (LD). LD is a statistical association between different genetic variants induced by the tendency of alleles that are close together on a chromosome to be inherited together and can occur when there is non-random association of variants at different loci. By this principle, two SNPs have LD if they are observed to be inherited together more often than expected. The likelihood of LD increases if the SNPs are located close to one another on the chromosome. For example, a SNP affecting the expression of gene A may be in linkage disequilibrium with a SNP that affects expression of gene B. If the product of gene B is causally related to the disease outcome, it would be wrong to conclude that gene A—or the dependent biomarker—is responsible for the phenotype, although such association could be found. As a result, to limit the influence of LD, ideal gene variants for Mendelian randomization are those not located in close proximity to other genes which also exert an influence on an outcome through alternate pathways.
A third limitation of Mendelian randomization is that precise estimates of causal effect are often biased. For example, causal effect estimates from Mendelian randomization studies can be thought of as a population-average effect (i.e., as if the intervention was applied to the entire population) and could be different than the effect of interventions applied to specific subgroups. On the other hand, weak genetic instruments, that explain too little variation in the exposure, could bias causal estimates or result in failure to establish causal relationships due do a lack of power. Use of large sample sizes or a genetic score combining multiple SNPs with additive or robust associations with the outcome of interest partially alleviate this concern. An additional caveat to interpretation of this technique is canalization. Canalization refers to the development of counter-regulatory mechanisms in response to a genetic variant. These mechanisms can take the form of genetic redundancy or alternative pathways that alter the relationship between a genetic variant and an outcome. For example, canalization can potentially be seen with variants associated with CYP1A1 gene which encodes for a cytochrome P450 enzyme. The highly inducible form of this enzyme is associated with an increased risk of lung cancer. In light smokers with this variant, the risk of lung cancer is increased by 7-fold. In heavy smokers with this variant, however, the risk of lung cancer is increased by 2-fold [9]. The differential impact of this variant may be the result of canalization through which the body’s response to heavy vs. light smoking complicates the interpretation of adverse effects of smoking on lung cancer development.
Mendelian Randomization in Cardiovascular Disease
Many studies have assessed the relationship between genetic variants, risk factors, and cardiovascular disease (CVD) (Table 1). One important risk factor that has been investigated using his methodology is obesity. Although obesity is frequently associated with CVD in observational studies, it is unclear if obesity causes CVD or if it is a confounder for other CVD risk factors. Using a composite of genetic variants associated with BMI, three studies have used Mendelian randomization to establish a potential causal link between BMI and CVD [13•, 23, 24]. Interestingly, a different study by Holmes et al. established a potential causal relationship between BMI and CVD risk factors, such as blood pressure, fasting glucose, inflammation, and type 2 diabetes mellitus (T2DM) but failed to show a similar relationship between obesity and CVD [25]. All of these studies have limitations including lack of specificity of the obesity phenotype and uncertainty regarding the presence of canalization. Abdominal, or central, obesity may be a more CVD-specific obesity phenotype. Observational studies have evaluated the association between abdominal adiposity (using waist-hip ratio [WHR] as a surrogate measure) and CVD with varying results [35,36,37]. Emdin et al. recently utilized a developed central obesity risk score comprised of 48 SNPs associated with WHR adjusted for BMI. The genetic risk score was positively associated with both T2DM and CVD outcomes. However, the score was also associated with alternative CVD risk factors such as triglyceride-rich lipoproteins, insulin, glucose, and systolic blood pressure. This alone does not invalidate the use of obesity variants as genetic instruments for testing the causal relationship between BMI or WHR and CVD. On the contrary, this suggests that alternative CVD risk factors could be direct consequences of obesity, rather than simply correlative factors. Similar findings were seen from a more recent Mendelian randomization study conducted by Dale et al. [13•]. Obesity has also been associated with atrial fibrillation in multiple observational studies and longitudinal exposure to obesity is associated with increased atrial fibrillation [38,39,40,41,42,43]. Chatterjee et al. used Mendelian randomization to evaluate the causal relationship between obesity and atrial fibrillation. Their study assessed the association between a genetic locus (FTO) which has been shown to have the strongest association with obesity and atrial fibrillation [44]. Additionally, they evaluated the association between a weighted composite of 39 SNPs associated with obesity and atrial fibrillation. Their results showed a positive association between obesity and atrial fibrillation using both methods, strengthening the evidence for a causal relationship [45•]. However, there were several caveats to this study, such as the association between the genetic variants and BMI was stronger at younger ages. Additionally, SNPs that were recently found to be associated with BMI were not included in this study.
Blood cholesterol levels are an established risk factor for CVD. While it has been accepted that LDL-C plays a causal role in the development of CVD, there are a number of unanswered questions regarding this relationship [10•]. Despite reductions in LDL-C, patients on statin therapy can continue to have CVD events. Using Mendelian randomization, genetic variants associated with lower LDL-C were used to assess the effect of long-term LDL-C reduction on CVD. They found that exposure to lower LDL-C earlier in life was protective in the development of CVD [11], suggesting early initiation of statin therapy may be beneficial. Mendelian randomization was also utilized to study the relationship between T2DM and LDL-C. Statin therapy has been associated with an increased risk of developing T2DM [46, 47]. However, it is unclear if the risk of T2DM is due to LDL-C levels or statin use. Using variants associated with LDL-C, White et al. found reduced risk of T2DM with higher LDL-C. This finding contributes to the evidence that lipid-lowering therapy may have unintended cardiometabolic consequences [12]. Mendelian randomization studies have also been employed to understand the relationship between triglycerides and CVD. Observational studies have shown a positive association between triglycerides and risk of CVD but RCTs of triglyceride-lowering medications have been negative [48, 49]. Two Mendelian randomization studies have been performed to better understand this discordance. Holmes et al. used a genetic risk score consisting of variants associated with triglyceride levels. They demonstrated that genetic predisposition for higher triglyceride levels is associated with a higher risk of CVD [10•]. Similar findings were demonstrated in a study performed by White et al. [12]. Mendelian randomization has also been used to assess the relationship between high-density lipoprotein cholesterol (HDL-C), cholesteryl ester transfer protein (CETP), and CVD. Inhibition of CETP is known to result in higher HDL-C levels. GWAS studies have also shown a correlation between polymorphisms in the CETP locus and variation in lipid levels. A meta-analysis using Mendelian randomization of the CETP polymorphism and coronary artery disease (CAD) risk has shown mixed results. One study showed no risk reduction with higher genetically determined HDL-C levels [21, 50,51,52,53]. In contrast, a second study utilizing this polymorphism showed a reduced risk of CVD with reduction in circulating genetically determined CETP [22]. Additional testing evaluating the relationship between HDL-C and risk of CVD has been inconclusive. Using genetic variants associated with HDL-C, two studies failed to show a causal association between HDL-C and CVD using Mendelian randomization [10•, 12].
Plasma levels of C-reactive protein (CRP) are independently associated with risk of coronary heart disease, but whether CRP is causally associated with coronary heart disease or merely a marker of underlying atherosclerosis is uncertain. As briefly mentioned above, Elliott et al. carried out a genome-wide association (n = 17,967) and replication study (n = 13,615) to identify genetic loci associated with plasma CRP concentrations. They then carried out a Mendelian randomization study of the most closely associated SNPs in the CRP locus and published data on other CRP variants involving a total of 28,112 cases and 100,823 controls, to investigate the association of CRP variants with coronary heart disease. Polymorphisms in five genetic loci were strongly associated with CRP levels. However, genetic variants in the CRP locus showed no association with coronary heart disease: OR, 1.00; 95% CI, 0.97 to 1.02. The lack of concordance between the effect on coronary heart disease risk of CRP genotypes and CRP levels therefore argues against a causal association of CRP with coronary heart disease [8••].
Many other biomarkers linked to CVD risk have been investigated using Mendelian randomization, including markers of metabolic regulation, hemodynamic stress, inflammation, hormones, and vitamin levels. For example, adiponectin is a protein secreted by mature adipocytes that is downregulated in obese individuals [54]. In several observational studies, low adiponectin was associated with insulin resistance, T2DM, and dyslipidemia [55,56,57]. However, the association between adiponectin and CVD has been variable [58,59,60,61]. Utilizing Mendelian randomization, Borges et al. evaluated the effect of SNPs associated with adiponectin levels on CVD. Their results did not show a relationship between genetically determined adiponectin levels and CVD [26]. Another example is N-terminal pro-B-type natriuretic peptide (NT-proBNP), a marker of hemodynamic stress and a prognostic biomarker in a variety of cardiac diseases [62,63,64,65,66]. GWAS have identified a SNP in the promoter of the natriuretic peptide precursor B gene associated with NT-proBNP levels. Using data from the PLATelet inhibition and patient Outcomes (PLATO) trial, no association was seen between the primary composite outcome of cardiovascular death, myocardial infarction, and stroke and genetically determined NT-proBNP levels [27]. This provides evidence that NT-proBNP may be a marker, rather than a mediator, in the etiological pathway to CVD. Many other biomarkers have been studied with mixed results (Table 1), although the clinical implication of many of these biomarkers for risk screening, prevention, and treatment remain unclear.
Conclusions
As the use of Mendelian randomization becomes increasingly common, understanding this method of clinical research has become ever more important. This technique has been used to evaluate the potential etiological role of emerging risk factors for cardiovascular disease. It has also been used in creative and interesting ways to address questions that would be difficult to answer in clinical trials such as whether genetic risk for coronary disease is modifiable by lifestyle [67]. However, accurate interpretation of the results of these and future studies will require a keen understanding of the advantages and disadvantages of this technique. As our understanding of genetics and genetic variants improve, the ability to identify additional risk loci for cardiovascular disease will increase, opening new avenues for investigation using Mendelian randomization. It is important to note that this technique is still inherently observational in nature and that the best method to answer many clinical questions, when feasible, is still a randomized controlled trial. While Mendelian randomization will remain an important tool in evaluating causality in epidemiologic research and provide further insight into risk factors for CVD, understanding the strengths and limitations of the technique is important for appropriate interpretation of results and drawing clinical conclusions.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Katan MB. Apolipoprotein E isoforms, serum cholesterol, and cancer. Lancet. 1986;1(8479):507–8.
Holmes MV, Ala-Korpela M, Smith GD. Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat Rev Cardiol. 2017;14(10):577–90. https://doi.org/10.1038/nrcardio.2017.78.
Smith GD, Ebrahim S. Mendelian randomization: prospects, potentials, and limitations. Int J Epidemiol. 2004;33(1):30–42. https://doi.org/10.1093/ije/dyh132.
Tate JR, Rifai N, Berg K, Couderc R, Dati F, Kostner GM, et al. International Federation of Clinical Chemistry standardization project for the measurement of lipoprotein(a). Phase I. Evaluation of the analytical performance of lipoprotein(a) assay systems and commercial calibrators. Clin Chem. 1998;44(8 Pt 1):1629–40.
Marcovina SM, Koschinsky ML, Albers JJ, Skarlatos S. Report of the National Heart, Lung, and Blood Institute Workshop on lipoprotein(a) and cardiovascular disease: recent advances and future directions. Clin Chem. 2003;49(11):1785–96. https://doi.org/10.1373/clinchem.2003.023689.
Kronenberg F, Trenkwalder E, Dieplinger H, Utermann G. Lipoprotein(a) in stored plasma samples and the ravages of time. Why epidemiological studies might fail. Arterioscler Thromb Vasc Biol. 1996;16(12):1568–72. https://doi.org/10.1161/01.ATV.16.12.1568.
Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331–9. https://doi.org/10.1001/jama.2009.801.
•• Elliott P, Chambers JC, Zhang W, Clarke R, Hopewell JC, Peden JF, et al. Genetic loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302(1):37–48. https://doi.org/10.1001/jama.2009.954. This study evaluated the relationship between several variants associated with CRP levels and risk of development of CVD. No association was seen between a variant in the CRP gene and risk of CVD; however, there was positive association with a variant in the APOE. Variants in the APOE gene exhibits pleiotropy, affecting LDL-C levels in addition to CRP. This study highlights the strengths and limitations of Mendelian randomization as a technique to establish causal relationships.
Perera FP. Environment and cancer: who are susceptible? Science. 1997;278(5340):1068–73. https://doi.org/10.1126/science.278.5340.1068.
• Holmes MV, Asselbergs FW, Palmer TM, Drenos F, Lanktree MB, Nelson CP, et al. Mendelian randomization of blood lipids for coronary heart disease. Eur Heart J. 2015;36(9):539–50. https://doi.org/10.1093/eurheartj/eht571. This study evaluated the role of LDL-C, triglycerides, and HDL-C on the development of CVD. Using unique genetic risk scores for each of these variables, they confirmed a causal relationship between both LDL-C and triglycerides and CVD. They were unable to show that HLD-C was causally related to the development with CVD, similar to the many published trials on HDL-C raising medications.
Ference BA, Yoo W, Alesh I, Mahajan N, Mirowska KK, Mewada A, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60(25):2631–9. https://doi.org/10.1016/j.jacc.2012.09.017.
White J, Swerdlow DI, Preiss D, Fairhurst-Hunter Z, Keating BJ, Asselbergs FW, et al. Association of lipid fractions with risks for coronary artery disease and diabetes. JAMA Cardiol. 2016;1(6):692–9. https://doi.org/10.1001/jamacardio.2016.1884.
• Dale CE, Fatemifar G, Palmer TM, White J, Prieto-Merino D, Zabaneh D, et al. Causal associations of adiposity and body fat distribution with coronary heart disease, stroke subtypes, and type 2 diabetes mellitus: a Mendelian randomization analysis. Circulation. 2017;135(24):2373–88. https://doi.org/10.1161/CIRCULATIONAHA.116.026560. This study examined the relationship between general obesity and central obesity and risk of CVD. The investigators developed a more comprehensive genetic risk score than prior studies and found a positive association between both general and central obesity and CVD. They also examined a number of CVD risk factors and intermediate markers of CVD such as ECG parameters and found positive associations with several of these variables. This study highlights the causal role of general and central obesity in the development of CVD and identified several possible biologic mechanisms.
Emdin CA, Khera AV, Natarajan P, Klarin D, Zekavat SM, Hsiao AJ, et al. Genetic association of waist-to-hip ratio with cardiometabolic traits, type 2 diabetes, and coronary heart disease. JAMA. 2017;317(6):626–34. https://doi.org/10.1001/jama.2016.21042.
Toutouzas K, Klettas D, Anousakis-Vlachochristou N, Melidis K, Azilazian Z, Asimomiti M, et al. The -174 G>C interleukin-6 gene polymorphism is associated with angiographic progression of coronary artery disease over a 4-year period. Hell J Cardiol. 2017;58(1):80–6. https://doi.org/10.1016/j.hjc.2017.02.002.
Zhao JV, Schooling CM. Endogenous androgen exposures and ischemic heart disease, a separate sample Mendelian randomization study. Int J Cardiol. 2016;222:940–5. https://doi.org/10.1016/j.ijcard.2016.07.174.
Gill D, Del Greco MF, Walker AP, Srai SKS, Laffan MA, Minelli C. The effect of iron status on risk of coronary artery disease: a Mendelian randomization study-brief report. Arterioscler Thromb Vasc Biol. 2017;37(9):1788–92. https://doi.org/10.1161/ATVBAHA.117.309757.
Schooling CM. Plasma levels of vitamin K and the risk of ischemic heart disease: a Mendelian randomization study. J Thromb Haemost. 2016;14(6):1211–5. https://doi.org/10.1111/jth.13332.
Merino J, Leong A, Posner DC, Porneala B, Masana L, Dupuis J, et al. Genetically driven hyperglycemia increases risk of coronary artery disease separately from type 2 diabetes. Diabetes Care. 2017;40(5):687–93. https://doi.org/10.2337/dc16-2625.
Zhan Y, Karlsson IK, Karlsson R, Tillander A, Reynolds CA, Pedersen NL, et al. Exploring the causal pathway from telomere length to coronary heart disease: a network Mendelian randomization study. Circ Res. 2017;121(3):214–9. https://doi.org/10.1161/CIRCRESAHA.116.310517.
Wu Z, Lou Y, Qiu X, Liu Y, Lu L, Chen Q, et al. Association of cholesteryl ester transfer protein (CETP) gene polymorphism, high density lipoprotein cholesterol and risk of coronary artery disease: a meta-analysis using a Mendelian randomization approach. BMC Med Genet. 2014;15(1):118. https://doi.org/10.1186/s12881-014-0118-1.
Niu W, Qi Y. Circulating cholesteryl ester transfer protein and coronary heart disease: Mendelian randomization meta-analysis. Circ Cardiovasc Genet. 2015;8(1):114–21. https://doi.org/10.1161/CIRCGENETICS.114.000748.
Hagg S, Fall T, Ploner A, Magi R, Fischer K, Draisma HH, et al. Adiposity as a cause of cardiovascular disease: a Mendelian randomization study. Int J Epidemiol. 2015;44(2):578–86. https://doi.org/10.1093/ije/dyv094.
Nordestgaard BG, Palmer TM, Benn M, Zacho J, Tybjaerg-Hansen A, Davey Smith G, et al. The effect of elevated body mass index on ischemic heart disease risk: causal estimates from a Mendelian randomisation approach. PLoS Med. 2012;9(5):e1001212. https://doi.org/10.1371/journal.pmed.1001212.
Holmes MV, Lange LA, Palmer T, Lanktree MB, North KE, Almoguera B, et al. Causal effects of body mass index on cardiometabolic traits and events: a Mendelian randomization analysis. Am J Hum Genet. 2014;94(2):198–208. https://doi.org/10.1016/j.ajhg.2013.12.014.
Borges MC, Lawlor DA, de Oliveira C, White J, Horta BL, Barros AJ. Role of adiponectin in coronary heart disease risk: a Mendelian randomization study. Circ Res. 2016;119(3):491–9. https://doi.org/10.1161/CIRCRESAHA.116.308716.
Johansson A, Eriksson N, Lindholm D, Varenhorst C, James S, Syvanen AC, et al. Genome-wide association and Mendelian randomization study of NT-proBNP in patients with acute coronary syndrome. Hum Mol Genet. 2016;25(7):1447–56. https://doi.org/10.1093/hmg/ddw012.
Zhao JV, Schooling CM. Homocysteine-reducing B vitamins and ischemic heart disease: a separate-sample Mendelian randomization analysis. Eur J Clin Nutr. 2017;71(2):267–73. https://doi.org/10.1038/ejcn.2016.246.
Kleber ME, Delgado G, Grammer TB, Silbernagel G, Huang J, Kramer BK, et al. Uric acid and cardiovascular events: a Mendelian randomization study. J Am Soc Nephrol. 2015;26(11):2831–8. https://doi.org/10.1681/ASN.2014070660.
Xu L, Lin SL, Schooling CM. A Mendelian randomization study of the effect of calcium on coronary artery disease, myocardial infarction and their risk factors. Sci Rep. 2017;7:42691. https://doi.org/10.1038/srep42691.
Leong A, Rehman W, Dastani Z, Greenwood C, Timpson N, Langsetmo L, et al. The causal effect of vitamin D binding protein (DBP) levels on calcemic and cardiometabolic diseases: a Mendelian randomization study. PLoS Med. 2014;11(10):e1001751. https://doi.org/10.1371/journal.pmed.1001751.
Manousaki D, Mokry LE, Ross S, Goltzman D, Richards JB. Mendelian randomization studies do not support a role for vitamin D in coronary artery disease. Circ Cardiovasc Genet. 2016;9(4):349–56. https://doi.org/10.1161/CIRCGENETICS.116.001396.
Zhong Y, Lin SL, Schooling CM. The effect of hematocrit and hemoglobin on the risk of ischemic heart disease: a Mendelian randomization study. Prev Med. 2016;91:351–5. https://doi.org/10.1016/j.ypmed.2016.09.003.
Svensson-Farbom P, Almgren P, Hedblad B, Engstrom G, Persson M, Christensson A, et al. Cystatin C is not causally related to coronary artery disease. PLoS One. 2015;10(6):e0129269. https://doi.org/10.1371/journal.pone.0129269.
Yusuf S, Hawken S, Ounpuu S, Bautista L, Franzosi MG, Commerford P, et al. Obesity and the risk of myocardial infarction in 27,000 participants from 52 countries: a case-control study. Lancet. 2005;366(9497):1640–9. https://doi.org/10.1016/S0140-6736(05)67663-5.
Vazquez G, Duval S, Jacobs DR Jr, Silventoinen K. Comparison of body mass index, waist circumference, and waist/hip ratio in predicting incident diabetes: a meta-analysis. Epidemiol Rev. 2007;29(1):115–28. https://doi.org/10.1093/epirev/mxm008.
Han TS, Bijnen FC, Lean ME, Seidell JC. Separate associations of waist and hip circumference with lifestyle factors. Int J Epidemiol. 1998;27(3):422–30. https://doi.org/10.1093/ije/27.3.422.
Azarbal F, Stefanick ML, Salmoirago-Blotcher E, Manson JE, Albert CM, LaMonte MJ et al. Obesity, physical activity, and their interaction in incident atrial fibrillation in postmenopausal women. J Am Heart Assoc. 2014;3(4). https://doi.org/10.1161/JAHA.114.001127.
Frost L, Hune LJ, Vestergaard P. Overweight and obesity as risk factors for atrial fibrillation or flutter: the Danish Diet, Cancer, and Health Study. Am J Med. 2005;118(5):489–95. https://doi.org/10.1016/j.amjmed.2005.01.031.
Huxley RR, Misialek JR, Agarwal SK, Loehr LR, Soliman EZ, Chen LY, et al. Physical activity, obesity, weight change, and risk of atrial fibrillation: the Atherosclerosis Risk in Communities study. Circ Arrhythm Electrophysiol. 2014;7(4):620–5. https://doi.org/10.1161/CIRCEP.113.001244.
Tedrow UB, Conen D, Ridker PM, Cook NR, Koplan BA, Manson JE, et al. The long- and short-term impact of elevated body mass index on the risk of new atrial fibrillation the WHS (women’s health study). J Am Coll Cardiol. 2010;55(21):2319–27. https://doi.org/10.1016/j.jacc.2010.02.029.
Vermond RA, Geelhoed B, Verweij N, Tieleman RG, Van der Harst P, Hillege HL, et al. Incidence of atrial fibrillation and relationship with cardiovascular events, heart failure, and mortality: a community-based study from the Netherlands. J Am Coll Cardiol. 2015;66(9):1000–7. https://doi.org/10.1016/j.jacc.2015.06.1314.
Wang TJ, Parise H, Levy D, D'Agostino RB Sr, Wolf PA, Vasan RS, et al. Obesity and the risk of new-onset atrial fibrillation. JAMA. 2004;292(20):2471–7. https://doi.org/10.1001/jama.292.20.2471.
Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–48. https://doi.org/10.1038/ng.686.
• Chatterjee NA, Giulianini F, Geelhoed B, Lunetta KL, Misialek JR, Niemeijer MN, et al. Genetic obesity and the risk of atrial fibrillation: causal estimates from Mendelian randomization. Circulation. 2017;135(8):741–54. https://doi.org/10.1161/CIRCULATIONAHA.116.024921. This study evaluated the causal role of obesity on the development of atrial fibrillation. They addressed this question using both a risk score consisting of multiple SNPS associated with BMI and the FTO gene which has the strongest association with BMI. Their results suggest a causal relationship between BMI and atrial fibrillation by both methods.
Preiss D, Seshasai SR, Welsh P, Murphy SA, Ho JE, Waters DD, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA. 2011;305(24):2556–64. https://doi.org/10.1001/jama.2011.860.
Swerdlow DI, Preiss D, Kuchenbaecker KB, Holmes MV, Engmann JE, Shah T, et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet. 2015;385(9965):351–61. https://doi.org/10.1016/S0140-6736(14)61183-1.
Emerging Risk Factors C, Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993–2000. https://doi.org/10.1001/jama.2009.1619.
Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366(9500):1849–61. https://doi.org/10.1016/S0140-6736(05)67667-2.
Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109–22. https://doi.org/10.1056/NEJMoa0706628.
Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367(22):2089–99. https://doi.org/10.1056/NEJMoa1206797.
Group HTRC, Bowman L, Hopewell JC, Chen F, Wallendszus K, Stevens W, et al. Effects of anacetrapib in patients with atherosclerotic vascular disease. N Engl J Med. 2017;377(13):1217–27. https://doi.org/10.1056/NEJMoa1706444.
Lincoff AM, Nicholls SJ, Riesmeyer JS, Barter PJ, Brewer HB, Fox KAA, et al. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med. 2017;376(20):1933–42. https://doi.org/10.1056/NEJMoa1609581.
Turer AT, Scherer PE. Adiponectin: mechanistic insights and clinical implications. Diabetologia. 2012;55(9):2319–26. https://doi.org/10.1007/s00125-012-2598-x.
Wildman RP, Mancuso P, Wang C, Kim M, Scherer PE, Sowers MR. Adipocytokine and ghrelin levels in relation to cardiovascular disease risk factors in women at midlife: longitudinal associations. Int J Obes. 2008;32(5):740–8. https://doi.org/10.1038/sj.ijo.0803782.
Li S, Shin HJ, Ding EL, van Dam RM. Adiponectin levels and risk of type 2 diabetes: a systematic review and meta-analysis. JAMA. 2009;302(2):179–88. https://doi.org/10.1001/jama.2009.976.
Yamamoto Y, Hirose H, Saito I, Nishikai K, Saruta T. Adiponectin, an adipocyte-derived protein, predicts future insulin resistance: two-year follow-up study in Japanese population. J Clin Endocrinol Metab. 2004;89(1):87–90. https://doi.org/10.1210/jc.2003-031163.
Zhang BC, Liu WJ, Che WL, Xu YW. Serum total adiponectin level and risk of cardiovascular disease in Han Chinese populations: a meta-analysis of 17 case-control studies. Clin Endocrinol. 2012;77(3):370–8. https://doi.org/10.1111/j.1365-2265.2011.04260.x.
Zhang H, Mo X, Hao Y, Huang J, Lu X, Cao J, et al. Adiponectin levels and risk of coronary heart disease: a meta-analysis of prospective studies. Am J Med Sci. 2013;345(6):455–61. https://doi.org/10.1097/MAJ.0b013e318262dbef.
Hao G, Li W, Guo R, Yang JG, Wang Y, Tian Y, et al. Serum total adiponectin level and the risk of cardiovascular disease in general population: a meta-analysis of 17 prospective studies. Atherosclerosis. 2013;228(1):29–35. https://doi.org/10.1016/j.atherosclerosis.2013.02.018.
Sook Lee E, Park SS, Kim E, Sook Yoon Y, Ahn HY, Park CY, et al. Association between adiponectin levels and coronary heart disease and mortality: a systematic review and meta-analysis. Int J Epidemiol. 2013;42(4):1029–39. https://doi.org/10.1093/ije/dyt087.
Jernberg T, James S, Lindahl B, Stridsberg M, Venge P, Wallentin L. NT-proBNP in unstable coronary artery disease—experiences from the FAST, GUSTO IV and FRISC II trials. Eur J Heart Fail. 2004;6(3):319–25. https://doi.org/10.1016/j.ejheart.2004.01.007.
Bibbins-Domingo K, Gupta R, Na B, Wu AH, Schiller NB, Whooley MA. N-terminal fragment of the prohormone brain-type natriuretic peptide (NT-proBNP), cardiovascular events, and mortality in patients with stable coronary heart disease. JAMA. 2007;297(2):169–76. https://doi.org/10.1001/jama.297.2.169.
Olsson LG, Swedberg K, Cleland JG, Spark PA, Komajda M, Metra M, et al. Prognostic importance of plasma NT-pro BNP in chronic heart failure in patients treated with a beta-blocker: results from the Carvedilol Or Metoprolol European Trial (COMET) trial. Eur J Heart Fail. 2007;9(8):795–801. https://doi.org/10.1016/j.ejheart.2007.07.010.
Wallentin L, Lindholm D, Siegbahn A, Wernroth L, Becker RC, Cannon CP, et al. Biomarkers in relation to the effects of ticagrelor in comparison with clopidogrel in non-ST-elevation acute coronary syndrome patients managed with or without in-hospital revascularization: a substudy from the Prospective Randomized Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation. 2014;129(3):293–303. https://doi.org/10.1161/CIRCULATIONAHA.113.004420.
Hijazi Z, Oldgren J, Andersson U, Connolly SJ, Ezekowitz MD, Hohnloser SH, et al. Cardiac biomarkers are associated with an increased risk of stroke and death in patients with atrial fibrillation: a Randomized Evaluation of Long-term Anticoagulation Therapy (RE-LY) substudy. Circulation. 2012;125(13):1605–16. https://doi.org/10.1161/CIRCULATIONAHA.111.038729.
Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med. 2016;375(24):2349–58. https://doi.org/10.1056/NEJMoa1605086.
Funding
Dr. Neeland is supported by grant K23DK106520 from the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institute of Health and by the Dedman Family Scholarship in Clinical Care from UT Southwestern.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Neeland reports personal fees from Boehringer-Ingelheim, American Heart Association, and from Advanced MR Analytics AB, plus grants from Novo Nordisk.
Dr. Savla has nothing to declare.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Novel and Emerging Risk Factors
Rights and permissions
About this article

Cite this article
Savla, J., Neeland, I.J. The Pros and Cons of Mendelian Randomization Studies to Evaluate Emerging Cardiovascular Risk Factors. Curr Cardiovasc Risk Rep 12, 2 (2018). https://doi.org/10.1007/s12170-018-0566-9
Published:
DOI: https://doi.org/10.1007/s12170-018-0566-9