
Abstract
Purpose
Simvastatin, a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, has antioxidant and anti-inflammatory properties that are independent of lipid-lowering abilities. This experiment was carried out to explore the effects of simvastatin on apoptosis in vulnerable atherosclerotic plaques of apoE-deficient mice.
Methods and results
Eight weeks-old apoE−/− mice were fed a Western-type diet. Vulnerable atherosclerotic lesions were formed in the branchiocephalic artery at the age of 30-weeks, before simvastatin administration for 8 weeks. Simvastatin did neither affect the levels of plasma glucose and lipids, nor the size of atherosclerotic lesions. Analysis of plaque composition showed that simvastatin decreased the area of lipid core and increased the amounts of macrophages and smooth muscle cells in atherosclerotic plaques of apoE−/− mice. In addition, simvastatin down-regulated the expression of vascular cell adhesion molecule-1 (VCAM-1) by both inhibition of nuclear factor kappa B (NF-кB) activation and suppression of the expression of the receptor for advanced glycation end products (RAGE). Moreover, we found that simvastatin administration led to reduced TUNEL-positive cells in the aortic root lesions, accompanied by up-regulation of Bcl-2 and Bcl-xL expression, and decreased P53 expression as shown by Western blot.
Conclusion
In the present study, we show novel data to suggest that simvastatin could suppress apoptosis in vulnerable atherosclerotic plaques of apoE−/− mice by regulating the expression of apoptosis-related proteins, such as p53, Bcl-2 and Bcl-xL.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rupture of the vulnerable atherosclerotic plaques is the major cause for fatal diseases such as acute coronary syndromes, myocardial infarction and strokes. Morphological analysis indicates that the vulnerable plaques are characterized by a large lipid-rich and collagen-poor core covered by a thin fibrous cap, a higher ratio of macrophages to vascular smooth muscle cells (VSMC). Especially the “necrotic core” (NC) or “lipid core” is a prominent feature of vulnerable plaques, which is predominantly caused by macrophage apoptosis [1].
Macrophages are the major cellular components of advanced plaques. They are full of lipids. Macrophage apoptosis leads to release of the debris and pro-inflammatory factors, which may further enhance macrophage death. Dead macrophages are ingested by neighboring phagocytes while the non-ingested apoptotic cells eventually become leaky and swollen, and then induce a process called “secondary necrosis” [2]. Phagocytic clearance of apoptotic macrophages, called efferocytosis, is deficient in advanced lesions, As a consequence, the dying macrophages are hardly cleared and the apoptotic macrophages eventually undergo secondary necrosis. The buildup of necrotic debris promotes inflammation, plaque instability, and acute thrombosis [3].
The apoptotic process is governed by pro-apoptotic and anti-apoptotic proteins. p53, a tumor-suppressor protein that has a the pro-apoptotic action, is inactivated and degraded under normal conditions, whereas the atherogenic stimuli, such as DNA damage, oxidative stress and oxidative lipoproteins, induce the expression of p53. Previous studies have found that p53 is co-localized with apoptotic macrophages in atherosclerotic plaques, and enhances the susceptibility of macrophages to apoptosis [4]. The elevated levels of p53 lead to pro-apoptotic responses by a combination of gene activation (e.g. p21WAF1 and Bax) and gene repression (e.g. Bcl-2) [5, 6]. Bcl-2, Bcl-xL and Bax are members of the Bcl-2 family, which are critical regulators of apoptosis in macrophages. Bcl-2 is a key survival molecule. Its protective role against macrophage apoptosis specifically in advanced lesions has been identified [7, 8]. Bax, the pro-apoptotic protein response for mitochondrial dysfunction and cell death, is essential for the death of macrophages in atherosclerotic plaques [9].
Simvastatin, a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, has anti-oxidant and anti-inflammatory properties that are independent of lipid-lowering abilities [10, 11]. However, the effects of simvastatin on apoptosis in vulnerable atherosclerotic plaques are not clear. In the present study, we show novel data that suggest that simvastatin could suppress apoptosis in vulnerable atherosclerotic plaques of apoE−/− mice by regulating the expression of apoptosis-related proteins, such as p53, Bcl-2 and Bcl-xL.
Methods
Preparation of animals
Male apoE−/− mice on a C57BL/6 J background were provided by Peking University Health Science Center (purchased from Jackson Laboratory). 8 Week-old apoE−/− mice (n = 30) were fed a Western-type diet containing 21% fat, 19.5% casein, and 0.21% cholesterol. Animals were kept at a temperature of 20–24°C and a humidity (45–55%)-controlled environment with a 12–12 h light-dark cycle. At 30 weeks of age, the apoE−/− mice were divided into a simvastatin group (n = 15) and a control group (n = 15). In the simvastatin group, simvastatin (25 mg/kg/day) was dissolved in aqueous methylcellulose and administered daily by oral garage at a dose of 25 mg/kg for 8 weeks, while the control group was treated with aqueous methylcellulose only. Blood samples were collected from the mice for measurement of serum glucose and lipids. All animal procedures were performed in accordance with the National Institutes of Health Animal Care and Use Guidelines. All animal protocols were approved by the Animal Ethics Committee at the Beijing Institute of Geriatrics.
Measurement of serum glucose and lipids
Serum glucose (GLU) levels were examined using the glucose oxidase method (Beckman). Total cholesterol (TC), triglycerides (TG), low-density lipoprotein-cholesterol (LDL-C) and high density lipoprotein-cholesterol (HDL-C) levels were measured using a kit from Sigma Diagnostics.
Quantification of atherosclerotic lesions in the branchiocephalic artery and aortic root
The hearts were sectioned throughout the aortic root with serial paraffin sections taken every 10 μm according to the identified methods [12]. The contiguous sections from the aortic root were stained with Movat pentachrome stain and the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labeling (TUNEL). The parts of the branchiocephalic arteries (BCA) were dissected and fixed overnight in 4% poly-merizedformaldehyde, followed by paraffin embedded, and sectioned 5-μm-thick. Contiguous sections were deparaffinized in xylene and rehydrated in graded series of ethanol. For area measurements and morphometric analysis, every fifth section was stained with a modified Movat pentachrome stain, Sirius Red, immunohistochemistry for MAC-3 and α-actin [13]. Plaque necrosis was quantified by measuring the area of Movat- Pentachrome negative cellular areas in the intima [14]. Collagen contents in the plaques were detected by polarized light after staining with Sirius Red. The atherosclerotic lesions were analyzed by the Image Pro-Plus-6 software (Media Cybernatics). In this experiment, the fibrous caps of many plaques in the control groups were broken or called ruptured, so the integrity of the fibrous cap was also evaluated. The plaque with broken fibrous cap was defined as such when it appeared broken in three BCA slides stained with Movat pentachrome.
Assessment of apoptosis in aortic root
To evaluate the apoptotic cells in the lesions of the aortic root, the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labeling (TUNEL; Promega) staining for 5 sections per mice, each separated by 60 μm, was carried out following the manufacturer’s instructions and the nuclei were counterstained with DAPI for 3 min before the slides were viewed by fluorescent microscopy (Carl Zeiss). The percentage of TUNEL-positive cells was calculated from the ratio of total nuclei in the whole plaque. Sections without the TdT enzyme were used as negative control.
Immunohistochemistry
Sections from BCA were used for immunohistochemistry staining to identify macrophages, smooth muscle cells (SMC) by a method reported previously [15]. Briefly, the sections were incubated with polyclonal antibodies at 37°C for 60 min or at 4°C overnight and then labeled with HRP-conjugated anti-rabbit IgG at 37°C for 60 min. Next, the coverslips were mounted with DABCO and analyzed by upright microscope (Carl Zeiss). Antibodies against MAC-3 (1:50 dilution) were purchased from BD Bioscience, α-actin (1:100 dilution) was purchased from Santa Cruz.
En face analysis of the descending aortas
Six descending aortas of each group were used for en face lipid staining. The aortas were dissected from the left subclavian artery to the iliac bifurcation, then opened longitudinally and stained with Oil Red O to visualize the extent of the lipid deposition. Quantitative analysis of lesion size was performed by first capturing aorta images with a Nikon D80. The data were analyzed with Image Pro-Plus-6 software.
Western blot
Descending arteries were dissected and used to analyze protein levels by Western blot. The lysates (10–30 μg protein) were separated by 10%SDS-PAGE, transferred to PVDF membrane (Millipore), blocked with 5% nonfat dry milk for 60 min, and probed overnight with antibodies at 4°C. The blots were incubated with HRP-conjugated anti-IgG for 1 h at 37°C, followed by detection with ECL (Santa Cruz). Antibodies against p53(1:1000 dilution), Bcl-2 (1:1000 dilution), Bax (Ser32) (1:500 dilution), VCAM-1 (1:1000 dilution), galectin-3/MAC-2(1:8000 dilution, a marker for macrophage) were purchased from Santa Cruz. Antibody to RAGE (1:500 dilution) was purchased from Sigma Aldrich. NF-кB p65 (1:1000 dilution) and phosphor-NFкB (Ser536) (1:500 dilution) and Bcl-xL (1:1000 dilution) were obtained from Cell Signaling, and CD68 (1:1000 dilution, another marker for macrophage) was purchased from AbD serotec.
Statistical analysis
All values are represented as means ± SE of the indicated number of measurements. One-way ANOVA test was used to determine significance, requiring P < 0.05 for statistical significance.
Results
Effect of simvastatin on the levels of serum glucose and serum lipid in apoE−/− mice
As shown in Table 1, no significant differences in body weight and levels of serum GLU, TC, TG, LDL-C and HDL-C were found between the simvastatin group and the control group.
Simvastatin did not affect atherosclerotic lesion size in apoE−/− mice
The percentage of aortic lesion area was measured by quantitative histomorphology of Oil Red O stained en face specimens. A similar percentage of aortic area (lesion area compared to total arch area) was affected after simvastatin administration (Fig. 1a). Similarly, no significant difference in volume fraction (volume lesion/volume vessel) of BCA and the aortic root was found between the simvastatin group and the control group, as shown by Movat staining (Fig. 1b and c). The results indicate that simvastatin did not affect vulnerable atherosclerotic lesion size in apoE−/− mice.

Simvastatin did not affect the atherosclerotic lesion size in apoE−/− mice. The percentage of aortic lesion area (lesion area compared to total arch area) was measured by quantitative histomorphology of Oil Red O stained en face specimens (a, n = 6). The volume fraction (volume lesion/volume vessel) of branchiocephalic arteries and aortic root was analyzed in the simvastatin and control groups, as shown by Movat staining (b and c, n = 6). Data represent the means ± SE
Simvastatin changed plaque composition in apoE−/− mice
The results indicate a significant decrease of necrotic core after simvastatin administration (Fig. 2a), but an increase of macrophage amounts in atherosclerotic lesions (Fig. 2b). SMC amounts were higher in plaques and SMC were mainly located in fibrous cap (Fig. 2c) after simvastatin administration, which facilitates to stabilize atherosclerotic plaques.

Simvastatin changed plaque composition in apoE−/− mice. The necrotic core from branchiocephalic arteries was detected by Movat staining (a). The amounts of macrophages (b) and SMC (c) in the plaques from branchiocephalic arteries were determined by immunohistochemistry. Data represent the means ± SE (n =6). * P < 0.05 versus control
Simvastatin decreased VCAM-1 expression through suppression of RAGE expression and NF-кB activation
As shown in Fig. 3, simvastatin administration resulted in a significant decrease of RAGE (Fig. 3a and b) and VCAM-1 (Fig. 3c) expression, suggesting the anti-inflammatory activity of simvastatin. As shown in Fig. 3c, simvastatin resulted in a reduced level of NF-кB and phosphorylation of NF-кB in descending arteries of apoE−/− mice, indicating that simvastatin could down-regulate the expression of adhesion molecules by both inhibition of NF-кB activation and suppression of RAGE expression.

Simvastatin decreased VCAM-1 expression through suppression of RAGE expression and NF-кB activation. The expression of RAGE in descending arteries and branchiocephalic arteries was measured by Western blot and immunohistochemistry (a, b). The levels of VCAM-1, NF-кB and phosphorylation of NF-кB in descending arteries of apoE−/− mice were determined by Western blot (c). Data represent the means ± SE (n = 4). * P < 0.05 versus control
Simvastatin regulated the expression of apoptosis-related proteins in atherosclerotic lesions
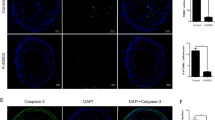
As shown in Fig. 4a, fewer TUNEL-positive cells appeared in the plaques of apoE−/− mice treated with simvastatin. However, simvastatin led to notably increased macrophages (galectin-3/MAC-2 and CD68 are two markers of macrophages) in the plaques of apoE−/− mice (Fig. 4b), suggesting that simvastatin suppressed the apoptosis of macrophages in atherosclerotic vulnerable lesions. Moreover, Western blot and immunohistochemistry showed that simvastatin down-regulated p53 expression and up-regulated expression of Bcl-2 and Bcl-xL, but not Bax, in descending arteries of apoE−/− mice (Fig. 4c and d).

Simvastatin regulated the expression of apoptosis-related proteins in atherosclerotic lesions. The apoptosis of macrophages in atherosclerotic lesions was measured by TUNEL (a). The expression of macrophage markers (galectin-3/MAC-2 and CD-68) in descending arteries in apoE−/− mice was detected by Western blot (b). The apoptosis-related proteins (p53, Bcl-2, Bcl-xL and Bax) in descending arteries or sections of branchiocephalic arteries were measured by immunohistochemistry (c) and Western blot (d). Data represent the means ± SE (n = 4). * P < 0.05 versus control
Discussion
In the present study, we provide evidence suggesting that simvastatin could suppress apoptosis in vulnerable atherosclerotic plaques of apoE−/− mice. In particular, our findings (1) suggest that simvastatin attenuates the incidence of apoptosis in plaques, which may suppress the expansion of the necrotic core in the vulnerable atherosclerotic plaques, (2) associate the suppression of p53 expression and up-regulation of Bcl-2 and Bcl-xL expression by simvastatin with anti-apoptotic effects of simvastatin in atherosclerotic plaques, (3) demonstrate an inhibitory effect of simvastatin on expression of RAGE and its downstream adhesion molecules.
To evaluate the effect of simvastatin on apoptosis in vulnerable atherosclerotic plaques, the apoE−/− mice were fed a Western-type diet (0.21% cholesterol contained in the diet) for 22 weeks to induce the formation of vulnerable plaques in BCA and then simvastatin was administrated for 8 weeks by orally. As known, plaque composition rather than plaque size determines its vulnerability. Therefore, we evaluated the composition and size of atherosclerotic plaques in apoE−/− mice treated with simvastatin. The results show that administration of the simvastatin reduced the size of lipid-rich necrotic core and increased the amounts of macrophages and SMC in atherosclerotic plaques. Adhesion molecules such as ICAM-1 and VCAM-1 are the most important cytokines responsible for the macrophage accumulation. Oxidative stress induces the expression of RAGE and then activates the NF-кB signal pathway which results in up-regulation of numerous cytokines [16]. RAGE is expressed in cells relevant to atherosclerosis, such as monocyte derived macrophages, endothelial cells, SMCs and lymphocytes, acting as a multiligands receptor to recognize high-mobility group box 1 (HMGB1), S100/Calgranulins and AGEs, which are significantly elevated in a hyperlipidemic and/or hyperglycaemic environment. RAGE has the unique ability to promote the activation of NF-κB, the most important transcriptional factor in atherosclerotic inflammation [17]. The interaction of ligands and RAGE causes the expression of many pro-atherosclerotic factors, including adhesion molecules, chemokines and pro-inflammatory cytokines. Although the RAGE-ligand axis provides an important role to accelerate atherosclerosis in diabetes, recent evidence highlights that it may contribute to chronic and amplified inflammatory status in atherosclerotic processes also beyond diabetes [18]. Previous studies have shown that RAGE deficiency inhibits the expression of pro-inflammatory adhesion molecules such as ICAM-1 and VCAM-1 in the hyperlipidaemic state, suggesting that RAGE is of pathophysiological importance under hyperlipidemic and normoglycemic state [19, 20]. In the present study, we also found that although simvastatin did not affect the levels of plasma glucose and down-regulated the expression of VCAM-1 by both inhibition of NF-кB activation and suppression of RAGE expression.
Macrophage infiltration often indicates inflammation. Macrophages are the major cellular components of advanced lesions and responsible for the secretion of inflammatory cytokines and proteolytic enzymes such as MMPs. These perpetuate the chronic inflammatory response and result in plaque expansion and degradation of the extracellular matrix leading to the lesion vulnerability/instability and eventually rupture. Suppression of adhesion molecule expression should lead to fewer macrophages recruited in the lesions. However, we found that are more macrophages and SMC were apparent in atherosclerotic plaques of apoE−/− mice treated with simvastatin. We speculate that these macrophages and SMC are resistant to apoptosis and survive after simvastatin treatment.
The vulnerable plaques are recognized by several histological characteristics, the most important features are the presence of a lipid-rich necrotic core, accompanied by an increased rate of apoptosis and defective efferocytosis. The dying/apoptotic macrophages may release lipid rich debris, inflammatory factors and proteases, and then induce secondary necrosis of neighboring cells. This process results in expansion of the lipid core, which contributes to the plaque instability to rupture and acute thrombosis [1, 2]. Research has revealed opposing roles for macrophage apoptosis in atherosclerotic plaque progression. In early lesions, macrophage apoptosis limits lesion cellularity and suppresses plaque progression. In advanced lesions, macrophage apoptosis promotes the development of the necrotic core, a key factor in rendering plaques vulnerable to disruption and to acute lumen thrombosis [3]. In the present study, we found decreased area of plaque necrosis and reduced TUNEL-positive cells in atherosclerotic plaques of apoE−/− mice treated with simvastatin. Moreover, it has been reported that statins significantly inhibit/suppress the inflammatory reaction induced by atheroma-associated macrophages. The macrophages treated with simvastatin become “harmless” and hardly destroy the stability of the vulnerable plaques [21]. Therefore, increased numbers of macrophages and SMC in the lesion of apoE−/− mice treated with simvastatin may contribute to lesion stabilization.
Lately, apoptosis, almost exclusively in macrophages and SMC, has been thought to be directly related to the destabilization/vulnerability of the advanced plaques. Although it has been reported that statins promote apoptosis of the cultured macrophages and SMC by special apoptotic stimuli in vitro, there is no evidence indicating enhanced apoptosis or cell death in animal atherosclerotic models after statin treatment. In the present study, we suggest that simvastatin has the ability to protect against the accumulation of apoptotic cells in the arterial wall. Simvastatin stabilized the advanced lesions at least in part by decreasing the incidence of apoptosis in plaques. This anti-apoptotic effect of simvastatin is of benefit in addition to its anti-inflammatory and anti-oxidant properties leading to the reduction of cytokines release and oxidative stress, independently of its cholesterol-lowering ability.
In order to further investigate the mechanism underlying the anti-apototic effects of simvastatin in atherosclerotic plaques, we analyzed the expression of the proapoptotic proteins Bax and p53 and the anti-apoptotic proteins Bcl-2 and Bcl-xL in the descending arteries. Western blot showed that simvastatin down-regulated p53 expression and up-regulated the expression of Bcl-2 and Bcl-xL, but not Bax. A previous study has shown that in atherosclerotic lesions, p53 co-localizes with apoptotic macrophages and that the apoptotic process of macrophages is also tightly regulated by this tumor suppressor protein [22]. It is suggested that p53 is up-regulated and activated by various stimuli, including DNA damage, oxidative stress and oxidized lipoproteins. In normal conditions, p53 is both inactivated and bound with MDM2, which blocks the transcriptional activation domain of p53 and promotes the ubiquitin-mediated modification or proteolysis and the proteasome-mediated degradation of p53. In contrast, activated p53 inhibits its own degradation and promotes p53 expression, which leads to anti-proliferative and pro-apoptotic responses by a combination of gene activation (e.g., p21WAF1 and Bax) and gene repression (e.g. Bcl-2) [5, 6]. In addition, the Bcl-2 family, consisting of pro-apoptotic (e.g. Bax and Bak) and anti-apoptotic proteins (e.g. Bcl-2 and Bcl-xL), are also involved in the apoptotic process. Their intracellular levels determine the overall sensitivity of the cell to apoptotic stimuli [8]. Edward Thorp and colleagues determined the effect of macrophage-targeted deletion of Bcl-2 on macrophage apoptosis in atherosclerotic lesions of ApoE−/− mice. Their results show that intimal cell apoptosis is increased in lesions of Bcl2flox-LysMCre;apoE−/− mice fed a Western diet for 10 weeks, but not 4 weeks, indicating that macrophage Bcl-2 might play a protective role against macrophage apoptosis, specifically in advanced atherosclerotic lesions of apoE−/− mice [7]. Moreover, in apoE−/− mice over-expressing human Bcl-2 in macrophages (Mø-hBcl-2 apoE−/− mice) and fed a Western diet for 15 weeks, TUNEL-positive cells in plaques from these mice were 3-fold less abundant than in control mice, suggesting a critical role in sustaining the survival of macrophages [8]. Bcl-2 and Bcl-xL have the ability to form heterodimers with pro-apoptotic proteins such as Bax and Bad, protecting cells from death. Previous studies have shown that Bcl-xL is even more important than Bcl-2 in protecting activated macrophages from death under particular apoptotic stimuli such as the LPS, NO and ROS which are abundant in atherosclerotic conditions [23]. Furthermore, several publications indicate that Bcl-xL is the main anti-apoptotic gene up-regulated upon differentiation of monocytes into macrophages [24, 25]. Also, Liu and coworkers showed that Bax deficiency reduces the apoptotic activity in macrophages [9]. Consistent with these studies, our results indicate that simvastatin down-regulates the expression of p53 and up-regulates the expression of antiapoptotic protein Bcl-2 and Bcl-xL, accompanied by fewer TUNEL-positive cells in atherosclerotic plaques of apoE−/− mice.
In conclusion, this study provides data that support our hypothesis that simvastatin has an anti-apoptotic effect and stabilizes the vulnerable atherosclerotic plaques mainly by suppression of the development of the necrotic core, and by regulating the expression of anti-and pro-apoptotic proteins, Bcl-2, Bcl-xL and p53, in apoE−/− mice.
References
Burke AP, Kolodgie FD, Farb A, Weber D, Virmani R. Morphological predictors of arterial remodeling in coronary atherosclerosis. Circulation. 2002;105:297–303.
Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol. 2009;86:1089–95.
Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res. 2009;50:S382–387.
Ihling C, Menzel G, Wellens E, Mönting JS, Schaefer HE, Zeiher AM. Topographical association between the cyclin-dependent kinases inhibitor P21, p53 accumulation, and cellular proliferation in human atherosclerotic tissue. Arterioscler Thromb Vasc Biol. 1997;17:2218–24.
von der Thüsen JH, van Vlijmen BJ, Hoeben RC, et al. Induction of atherosclerotic plaque rupture in apolipoprotein E−/− mice after adenovirus-mediated transfer of p53. Circulation. 2002;105:2064–70.
Tabas I. p53 and atherosclerosis. Circ Res. 2001;88:747–9.
Thorp E, Li Y, Bao L, et al. Brief report: increased apoptosis in advanced atherosclerotic lesions of Apo E−/− mice lacking macrophage Bcl-2. Arterioscler Thromb Vasc Biol. 2009;29:169–72.
Gautier EL, Huby T, Witztum JL, et al. Macrophage apoptosis exerts divergent effects on atherogenesis as a function of lesion stage. Circulation. 2009;119:1795–804.
Liu J, Thewke DP, Su YR, Linton MF, Fazio S, Sinensky MS. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler Thromb Vasc Biol. 2005;25:174–9.
Wierzbicki AS, Poston R, Ferro A. The lipid and non-lipid effects of statins. Pharmacol Ther. 2003;99:95–112.
Marzilli M. Pleiotropic effects of statins: evidence for benefits beyond LDL-cholesterol lowering. Am J Cardiovasc Drugs. 2010;10:S3–9.
Zuckerman SH, Evans GF, Schelm JA, Eacho PI, Sandusky G. Estrogen-mediated increases in LDL cholesterol and foam cell-containing lesions in human ApoB100xCETP transgenic mice. Arterioscler Thromb Vasc Biol. 1999;19:1476–83.
Bea F, Blessing E, Bennett B, Levitz M, Wallace EP, Rosenfeld ME. Simvastatin promotes atherosclerotic plaque stability in apoE-deficient mice independently of lipid lowering. Arterioscler Thromb Vasc Biol. 2002;22:1832–7.
Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe−/− mice. Arterioscler Thromb Vasc Biol. 2008;28:1421–8.
Tekabe Y, Li Q, Rosario R, et al. Development of receptor for advanced glycation end products-directed imaging of atherosclerotic plaque in a murine model of spontaneous atherosclerosis. Circ Cardiovasc Imaging. 2008;1:212–9.
Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605.
Bierhaus A, Schiekofer S, Schwaninger M, et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-кB. Diabetes. 2001;50:2792–808.
Basta G. Receptor for advanced glycation endproducts and atherosclerosis: From basic mechanisms to clinical implications. Atherosclerosis. 2008;196:9–21.
Sun L, Ishida T, Yasuda T, et al. RAGE mediates oxidized LDL-induced pro-inflammatory effects and atherosclerosis in non-diabetic LDL receptor-deficient mice. Cardiovasc Res. 2009;82:371–81.
Harja E, Bu DX, Hudson BI, et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J Clin Invest. 2008;118:183–94.
Yan ZQ, Hansson GK. Innate immunity, macrophage activation, and atherosclerosis. Immunol Rev. 2007;219:187–203.
Merched AJ, Williams E, Chan L. Macrophage-specific p53 expression plays a crucial role in atherosclerosis development and plaque remodeling. Arterioscler Thromb Vasc Biol. 2003;23:1608–14.
Okada S, Zhang H, Hatano M, Tokuhisa T. A physiologic role of Bcl-xL induced in activated macrophages. J Immunol. 1998;160:2590–6.
Chatterjee D, Han Z, Mendoza J, et al. Monocytic differentiation of HL-60 promyelocytic leukemia cells correlates with the induction of Bcl-xL. Cell Growth Differ. 1997;8:1083–9.
Antignani A, Youle RJ. The cytokine, granulocyte-macrophage colony-stimulating factor (GM-CSF), can deliver Bcl-XL as an extracellular fusion protein to protect cells from apoptosis and retain differentiation induction. J Biol Chem. 2007;282:11246–54.
Funding
This work was supported by grants from National Basic Research Program of China (2012CB517502) and National Natural Science Foundation of China (81070634).
Conflict of interest
None declared.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Weiwei Qin, Yonggang Lu, and Chengyan Zhan contributed equally to this work.
Rights and permissions
About this article
Cite this article
Qin, W., Lu, Y., Zhan, C. et al. Simvastatin Suppresses Apoptosis in Vulnerable Atherosclerotic Plaques Through Regulating the Expression of p53, Bcl-2 and Bcl-xL. Cardiovasc Drugs Ther 26, 23–30 (2012). https://doi.org/10.1007/s10557-011-6347-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-011-6347-z