
Abstract
In this study, we investigated the protective role of Mzb1 in atherosclerotic plaque vulnerability. To explore the impact of Mzb1, we analyzed Mzb1 expression, assessed apoptosis, and evaluated mitochondrial function in atherosclerosis (AS) mouse models and human vascular smooth muscle cells (HVSMCs). We observed a significant decrease in Mzb1 expression in AS mouse models and ox-LDL-treated HVSMCs. Downregulation of Mzb1 increased ox-LDL-induced apoptosis and cholesterol levels of HVSMCs, while Mzb1 overexpression alleviated these effect. Mzb1 was found to enhance mitochondrial function, as evidenced by restored ATP synthesis, mitochondrial membrane potential, and reduced mtROS production. Moreover, Mzb1 overexpression attenuated atherosclerotic plaque vulnerability in ApoE−/− mice. Our findings suggest that Mzb1 overexpression regulates the AMPK/SIRT1 signaling pathway, leading to the attenuation of atherosclerotic plaque vulnerability. This study provides compelling evidence for the protective effect of Mzb1 on atherosclerotic plaques by alleviating apoptosis and modulating mitochondrial function in ApoE−/− mice.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human atherosclerotic plaques are characterized by fibrofatty lesions within arterial walls. These plaques tend to rupture, leading to their entry into the circulation. Such plaque ruptures are widely believed to play a significant role in the development of heart attacks and strokes. Studies have shown that ruptured plaques are often characterized by a thin fibrous cap with high macrophage infiltration, covering a large necrotic core [1, 2]. The continuous accumulation of cytotoxic lipids in plaque tissue activates a malignant process of cell death, inflammation, and fibrous tissue degradation, ultimately weakening the fibrous cap and causing plaque rupture [3]. Despite extensive research on the pathological processes and identification of multiple risk factors for atherosclerotic plaque vulnerability, the mechanisms of plaque rupture remain poorly understood.
Vascular smooth muscle cells (VSMCs) are the primary cell type found in all atherosclerotic plaques [4]. In the process of disease progression, VSMCs can switch to different phenotypes, including phenotypes similar to foam cells, macrophages, mesenchymal stem cells and osteochondrocytes, which exert both positive and negative effects on disease progression [5, 6]. In early atherosclerosis, the VSMC uptake of oxidized low-density lipoprotein (ox-LDL) leads to foam cell formation and cell apoptosis. Additionally, activated VSMCs secrete chemokines, contributing to monocyte recruitment. In the late phase of atherosclerosis, VSMCs play a role in generating fibrous caps and forming necrotic cores, thereby increasing atherosclerotic plaque vulnerability [3, 6].
Mitochondria, vital cellular organelles responsible for energy production via oxidative phosphorylation, play crucial roles in cellular metabolism, calcium homeostasis, reactive oxygen species (ROS) production, and cell signaling. Proper mitochondrial function is essential for overall cellular health, especially in cells with high energy demand. Mitochondrial dysfunction is closely associated with various cardiovascular diseases, including ischemic heart disease, atherosclerosis, cardiomyopathy, and hypertension [7]. This dysfunction manifests as reduced ATP production, impaired mitochondrial regulation, increased generation of ROS and associated signaling pathways, dysregulated cell growth and apoptosis, compromised activity of the mitochondrial electron transport chain, and an inflammatory response [8,9,10]. Mitochondrial dysfunction is a key factor in foam cell formation and is a characteristic feature of atherosclerosis. Studies have demonstrated that ox-LDL, a major contributor to the development of atherosclerosis, promotes oxidative stress and subsequent mitochondrial dysfunction[11]. Accumulating studies have revealed that impaired mitochondrial function leads to increased oxidative stress, inflammation, and apoptosis within atherosclerotic plaques [12, 13]. However, mitochondrial abnormalities or dysfunctions can trigger apoptosis, thereby increasing the risk of plaque rupture [14]. Understanding the intricate relationship between mitochondrial function and plaque stability may reveal potential therapeutic targets to prevent plaque progression and reduce the risk of acute cardiovascular events.
Mzb1 (marginal zone B and B1 cell-specific protein), also referred to as plasma cell-induced resident endoplasmic reticulum protein (pERp1), is an 18 kDa protein localized within the endoplasmic reticulum (ER) [15, 16]. Many recent studies on Mzb1 have mainly focused on autoimmune diseases, periodontitis, and cancers [17,18,19]. In a study conducted by Wu et al., it was discovered that overexpression of Mzb1 enhanced apoptosis in rectal adenocarcinoma cells, leading to a higher rate of cell death [20]. One study reported that Mzb1 improved mitochondrial function in cardiomyocytes after myocardial infarction [21]. However, the association of Mzb1 with atherosclerotic plaque vulnerability has not yet been investigated. Therefore, we hypothesized that Mzb1 may play a role in attenuating atherosclerotic plaque vulnerability by regulating mitochondrial function and oxidative stress.
Methods
Animal Models
The in vivo study protocol was approved by the Ethics Committee of Chedun Experimental Animal Breeding Farm Co., Ltd. and conformed to the Guide for the Care and Use of Laboratory Animals published by the NIH (NIH publication, eighth edition, updated 2011). Eight-week-old male ApoE−/− mice were purchased from Charles River Laboratories (Beijing, China). Mice were randomly assigned to four groups (n = 8 per group): sham, Mzb1-oe (Mzb1 overexpression), HFD, and HFD + Mzb1-oe groups. The animals had free access to water during the treatment. Mice in the sham and Mzb1-oe groups were fed normal chow. Mice in the HFD and HFD + Mzb1-oe groups were fed an HFD (D12109C, Research Diets) for 12 weeks. Mzb1-oe lentivirus was intravenously injected into the mice every 3 weeks (1 × 107 TU/mL) during the 12 weeks, and then all mice were sacrificed and aortic tissue was collected for further analysis.
Evaluation of Atherosclerotic Lesion Development
The circulatory system was rinsed with phosphate buffered saline (PBS) and fixed with 4% paraformaldehyde. To prepare for further analysis, the heart and ascending aorta were embedded in an optimal cutting temperature (OCT) compound and rapidly frozen by placing them in liquid nitrogen. For the examination of lesion formation in the aortic roots, consecutive cross-sections with a thickness of 7 μm were collected from the area where the aortic valve leaflets originated. To detect lipid deposition, sections were stained with Oil Red O. The areas occupied by atherosclerotic lesions were quantified using the ImageJ software.
Histological Examination
Frozen sections of atherosclerotic lesions in the aortic roots were used for histological examination. The sections were stained with hematoxylin and eosin to visualize and assess the size of the necrotic core, and Masson’s staining was employed to determine the collagen content within the lesions.
Immunohistochemistry
For immunohistochemical staining, sections were initially treated with 3% hydrogen peroxide to neutralize endogenous peroxidase activity. Subsequently, the sections were incubated overnight at 4 °C with primary antibodies. The detection of macrophages and VSMCs involved the use of rabbit anti-mouse CD68 antibody (Proteintech, China) and rabbit anti-mouse α-SMA antibody (Proteintech, China), respectively. Microscopic images of the stained sections were captured using a Leica microscope, and these images were further analyzed and quantified using Image-Pro Plus software.
Cell Culture and Treatments
Human vascular smooth muscle cells (HVSMCs) were sourced from Zhong Qiao Xin Zhou Biotechnology Co., Ltd. (Shanghai, China), and ox-LDL (20605ES05) was obtained from Shanghai Yeasen BioTechnologies Co. Ltd. HVSMCs were cultured in smooth muscle cell medium (Cat No:1101; ScienCell), supplemented with 2% fetal bovine serum (FBS), 1% penicillin/streptomycin, and smooth muscle cell growth supplement (Cat No. 1152; ScienCell). Cells were incubated at 37 °C with 5% CO2 in a CO2 incubator to maintain the optimal conditions. Upon reaching 70%-80% confluence, HVSMCs were stimulated in smooth muscle cell medium mixed with 100 μg/mL ox-LDL for 48 h. Small interfering RNA against Mzb1(si-Mzb1), the Mzb1-oe, and their negative controls were obtained from GenePharma (Shanghai, China) and transfected into cells using Lipofectamine™ 3000 (Invitrogen, Carlsbad, CA, USA).
Determination of Cholesterol Levels
The levels of total cholesterol (TC), triglyceride (TG) and low-density lipoprotein cholesterol (LDL-C) in HVSMCs were detected by total cholesterol assay kit, triglyceride assay kit and low-density lipoprotein cholesterol assay kit, respectively (Nanjing Jiancheng Bioengineering Institute, China) according to the manufacturer’s instructions.
Western Blotting Analysis
Aorta samples and HVSMCs were lysed using radioimmunoprecipitation assay (RIPA) lysis buffer (EpiZyme, Shanghai, China) for 10 min, supplemented with PSMF and protein phosphatase inhibitors. The protein concentration was determined using a BCA Protein Assay Kit (EpiZyme, ZJ101), and protein loading buffer was added to the lysates at a ratio of 4:1 and thoroughly mixed. Thereafter, 10 μg of protein was loaded onto 10% or 15% SDS-PAGE gels for electrophoretic separation. The separated proteins were subsequently transferred onto 0.22 μm polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA), which were then blocked with Protein Free Rapid Blocking Buffer (EpiZyme, PS108P) for 10 min. After blocking, the membranes were incubated overnight at 4 °C with the designated primary antibodies, followed by incubation with the appropriate secondary antibody at 37 °C for 1 h. The immunoblotted membranes were scanned and detected using a calibrated densitometer (Bio-Rad). The intensity of the immunoblot bands was normalized to the loading control (β-actin or GAPDH). The following primary antibodies were employed in this study: Mzb1 (Invitrogen, 1:1000); Bax (Proteintech, 1:2000); Bcl 2 (Proteintech, 1:2000); Cytochrome c (Proteintech, 1:2000); AMPK (Proteintech, 1:1000); SIRT1 (Abcam, 1:1000); β-actin (Servicebio, 1:1000).
Quantitative RT–PCR
RT-qPCR was performed according to the manufacturer's instructions. Initially, total RNA was extracted using RNAiso Plus (Takara, Tokyo, Japan). Subsequently, 500 ng of RNA underwent reverse transcription using the Hifair® III 1st Strand cDNA Synthesis SuperMix (Yeasen Biotech Company, Shanghai, China). For qRT–PCR, the cDNA sample was used in combination with SYBR Green Master Mix (Yeasen Biotech Company) and the CFX96 Real-Time PCR System (Bio-Rad, Hercules, CA, USA).
Detection of Apoptosis using Flow Cytometry
The apoptosis rate was evaluated using an Annexin V-FITC/PI Cell Apoptosis Detection Kit (Servicebio) according to the manufacturer’s instructions. The cells were seeded into 6-well tissue culture plates. Following treatment, the cells were collected, washed with PBS, and resuspended in pre-cooled binding buffer, and the cell concentration was adjusted to 1 × 106 cells/mL. Subsequently, 5 μL of Annexin V-FITC and 5 μL of PI were added to 100 μL of cell suspension and kept away from light at room temperature for 10 min. Thereafter, 400 μL of binding buffer was added, the mixture was gently shaken, and flow cytometry analysis (BD FACSCanto) was performed within 1 h.
Transmission Electron Microscopy
The mice were anesthetized and perfused with a 0.9% saline solution. Aortic tissue blocks measuring 1 × 2 × 3 mm were carefully extracted. These fresh tissue blocks were subsequently placed in a fixative for transmission electron microscopy (TEM, Servicebio) and kept at 4 °C for 4 h. Thereafter, they were fixed in a 1% OsO4 solution in 0.1 mol/L PBS for 2 h at room temperature. Tissue blocks were dehydrated using an alcohol gradient. Afterward, the sections were embedded by baking in an oven set at 60 °C for 48 h and subsequently sliced into ultrathin Sects. (60 nm) using an ultramicrotome. Finally, the mitochondrial ultrastructure of the aorta was examined using a transmission electron microscope (HITACHI, HT7700, Japan).
ATP Determination
The intracellular ATP content of the HVSMCs was determined using an ATP assay kit (Beyotime). HVSMCs (1 × 106) were lysed and centrifuged at 4 ℃. After removing the cell pellets, the supernatant (20 μL each well) was transferred to black 96-well plates (Thermo Scientific, Waltham, MA, USA) containing ATP working solution (100 μL each well). Luminescence was recorded using a microplate reader (Thermo Scientific, Lithuania), and the ATP concentration was further divided by the protein concentration of the cells, as detected using the bicinchoninic acid assay (Beyotime).
JC-1 Measurements
The JC-1 Mitochondrial Membrane Potential Assay Kit (Servicebio, China) was used for detecting mitochondrial membrane potential following the manufacturer’s protocol (Servicebio, China). Fluorescence signals were measured at 514/529 nm (excitation/emission, green) and 585/590 nm (excitation/emission, red) using a high-resolution confocal laser-scanning microscope, FV1000 (Olympus, Germany). JC-1 accumulates in mitochondria, which is dependent on the mitochondrial membrane potential. In cells with a high mitochondrial membrane potential, JC-1 forms a complex emitting red fluorescence. Conversely, in cells with a low mitochondrial membrane potential, JC-1 remains in its monomeric form, resulting in green fluorescence.
Determination of Mitochondrial ROS Levels
The mitochondrial ROS (mtROS) levels in HVSMCs were measured using MitoSOX (Invitrogen, Carlsbad, CA, USA). HVSMCs were collected and incubated in medium containing 5 μM MitoSOX for 30 min at 37℃ in the dark. After incubation, the cells were washed twice with PBS, and the relative fluorescence intensity of MitoSOX was recorded using a high-resolution confocal laser scanning microscope FV1000 (Olympus, Germany). The excitation (EX) and emission wavelengths (EM) of MitoSOX were 510 nm and 580 nm, respectively.
Data Analysis
All data were presented as the mean ± standard deviation. Statistical analyses were conducted using GraphPad Prism software (version 9.0; GraphPad Software, Inc.). The significance of the differences between the two groups was determined using unpaired two-tailed Student's t-tests, with statistical significance set at p < 0.05.
Results
Downregulation of Mzb1 in Atherosclerotic Plaques and Ox-LDL-Treated HVSMCs
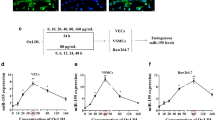
To investigate the role of Mzb1 in atherosclerotic plaques, we examined its expression in mice with atherosclerotic plaques. As shown in Fig. 1a-c, the protein and mRNA levels of Mzb1 were significantly lower in ApoE−/− mice with atherosclerotic plaques than in the sham group. Immunohistochemical staining for Mzb1 corroborated these findings (Fig. 1d, e). Similarly, a significant downregulation of Mzb1 was observed at both the protein (Fig. 1f, g) and mRNA (Fig. 1h) levels in primary cultured HVSMCs exposed to 100 μg/mL ox-LDL for 48 h.

Mzb1 expression levels are decreased in atherosclerotic plaques in ApoE−/− mice and ox-LDL-induced HVSMCs. Protein levels (a, b) and mRNA levels (c) of Mzb1 were detected using western blotting and RT-qPCR, respectively. Representative immunohistochemical images of aortic roots from ApoE.−/− mice (d, e). Scale bars: 200 μm. The expression of Mzb1 in HVSMCs stimulated with ox-LDL (100 μg/ml) for 48 h was detected using western blot (f, g) and RT-qPCR (h). Data are presented as the mean ± SD (n = 3). **p < 0 .01
Mzb1 Protects Against Ox-LDL-Induced HVSMCs Cholesterol Deposition and Apoptosis
To understand the impact of Mzb1 downregulation in ox-LDL-induced HVSMCs, we investigated changes in cell cholesterol deposition and apoptosis following the targeted suppression of Mzb1 using siRNA (si-Mzb1). The effectiveness of si-Mzb1 in silencing Mzb1 was confirmed by a significant reduction in Mzb1 protein levels (Fig. 2a, b). Ox-LDL increased the concentration of intracellular TC, TG and LDL-C. These increase was further exacerbated by si-Mzb1 treatment (Fig. 2c-e). As shown in Fig. 2f and g, flow cytometric analysis demonstrated a significant increase in the apoptosis rate 48 h after exposure to ox-LDL, and this increase was further exacerbated by si-Mzb1 treatment. To explore whether the increase in apoptosis was mediated through the mitochondrial death pathway, we examined changes in the expression of mitochondria-related factors, Bax, Bcl-2, and cytochrome c (cyt-c). Western blot analysis revealed that the protein levels of Bcl-2 decreased, whereas those of Bax increased upon si-Mzb1 treatment. Additionally, cyt-c expression was upregulated after Mzb1 silencing (Fig. 2h-k).

Mzb1 knockdown promoted HVSMC apoptosis. The efficiency of si-Mzb1 in HVSMCs was verified using western blot (a, b). TC, TG and LDL-C were measured in HVSMCs (c-e). The effects of si-Mzb1 in promoting HVSMCs apoptosis were assessed using flow cytometry (f, g). The impact of si-Mzb1 on HVSMCs apoptosis-related protein levels, including Bax, Bcl-2 and Cyt-c (h–k). Data are presented as the mean ± SD (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001;****p < 0.0001
Next, we examined the effects of Mzb1-oe on HVSMCs cholesterol deposition and apoptosis. Western blot analysis (Fig. 3a, b) revealed that the introduction of a plasmid carrying the Mzb1 gene significantly enhanced Mzb1 protein levels. Ox-LDL increased the concentration of intracellular TC, TG and LDL-C, and Mzb1-oe effectively counteracted these detrimental effects (Fig. 3c-e). Mitochondria are highly sensitive to various cellular environmental cues and initiate apoptotic pathways upon exposure to oxidative stress [22]. One critical aspect of this pathway is the abnormal upregulation of Bax and simultaneous downregulation of Bcl-2, leading to the subsequent release of Cyt-c from the mitochondria to the cytoplasm. Our findings indicated that ox-LDL significantly increased the protein levels of Cyt-c and Bax, indicating the activation of the mitochondrial death pathway. However, Mzb1-oe effectively counteracted these detrimental effects (Fig. 3f-i). Furthermore, flow cytometry results demonstrated that Mzb1-oe successfully attenuated ox-LDL-induced apoptosis of HVSMCs (Fig. 3j, k).

Mzb1 overexpression inhibited HVSMC apoptosis. The efficiency of MZB1 overexpression in HVSMCs was verified using western blot (a, b). TC, TG and LDL-C were measured in HVSMCs (c-e). The effects of Mzb1 overexpression on the HVSMC apoptosis-related protein levels of Bax, Bcl-2 and Cyt-c (f-i). The inhibitory effects of Mzb1 overexpression on HVSMCs apoptosis, as revealed by flow cytometry (j, k). Data are presented as the mean ± SD (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001;****p < 0.0001
Mzb1 Improves Mitochondrial Function
Mitochondrial damage, primarily induced by the excessive production of oxygen free radicals, is considered the main factor contributing to AS. In this study, we examined the impact of Mzb1 on mitochondrial membrane potential through confocal microscopic analysis of JC-1 staining (Fig. 4a, b). Exposure to ox-LDL resulted in the depolarization of the mitochondrial membrane potential, as evidenced by intensified JC-1 staining. This depolarizing effect induced by ox-LDL was further intensified in the presence of si-Mzb1. Since oxidative phosphorylation governs ATP production in mitochondria, we examined the potential influence of Mzb1 on ATP levels. Notably, when HVSMCs were exposed to 100 μg/mL ox-LDL for 48 h, a significant decrease in mitochondrial ATP levels was observed. Furthermore, the knockdown of Mzb1 amplified this detrimental change (Fig. 4c). We also examined the effect of Mzb1 on mtROS production after stimulation with ox-LDL. Confocal microscopy revealed a significant increase in mtROS levels after 48 h of ox-LDL stimulation, and si-Mzb1 exacerbated oxidative stress (Fig. 4d, e). Conversely, Mzb1 overexpression successfully countered the depolarization of the mitochondrial membrane potential (Fig. 4f, g). Enhanced Mzb1 function led to an elevation in ATP production even in the presence of ox-LDL (Fig. 4h). Increased mtROS production induced by ox-LDL was mitigated by Mzb1 overexpression (Fig. 4i, j).

Mzb1 significantly improves mitochondrial function in HVSMCs. JC-1 staining revealed that si-Mzb1 aggravated ox-LDL-induced depolarization of the mitochondrial membrane potential in HVSMCs (scale bar: 20 μm) (a). Statistical data for the fluorescence intensities determined through JC-1 staining (b). si-Mzb1 decreased the cellular ATP content in ox-LDL-induced HVSMCs (c). si-Mzb1 increases mitochondrial ROS levels in HVSMCs, as detected using MitoSOX staining (red, scale bar: 100 μm) (d). Quantification of mtROS levels (reflected by the relative fluorescence intensity of MitoSOX) (e). Mzb1-oe rescues ox-LDL-induced depolarization of the mitochondrial membrane potential in HVSMCs, as revealed by JC-1 staining (scale bar: 20 μm) (f). Statistical data for the fluorescence intensities determined using JC-1 staining (g). Mzb1-oe increased the cellular ATP content in ox-LDL-induced HVSMCs (h). Mzb1-oe suppresses mitochondrial ROS levels in HVSMCs, as detected using MitoSOX staining (red, scale bar: 100 μm) (i). Quantification of mtROS levels (reflected by the relative fluorescence intensity of MitoSOX) (j). n = 3, *p < 0.05; **p < 0.01; ***p < 0.001;****p < 0.0001
Mzb1 Attenuates Atherosclerotic Plaque Vulnerability in ApoE−/−Mice
Our investigation focused on examining the impact of Mzb1 on the stability of atherosclerotic plaques in mice. To assess plaque vulnerability, we employed histological and immunohistochemical (IHC) staining techniques to monitor stability in the aortic roots. Initially, a significant increase in lesion areas within plaques of ApoE−/− mice fed HFD was observed. However, the overexpression of Mzb1 rescued lesion areas (Fig. 5a, b). H&E staining revealed a significantly reduced necrotic core size in Mzb1-overexpressing mice compared to ApoE−/− mice (Fig. 5a, c). Furthermore, Masson staining demonstrated increased collagen content within the atherosclerotic plaques of Mzb1-overexpressing mice compared to those of ApoE−/− mice (Fig. 5a, d). To assess the impact of Mzb1 overexpression on plaque vulnerability, we examined the presence of CD68-positive macrophages associated with plaque instability using IHC staining. The findings revealed that Mzb1 overexpression significantly reduced the area occupied by CD68-positive macrophages within the plaque (Fig. 5a, e). Considering the crucial role of extracellular matrix proteins, such as collagen, in the formation of a stable fibrous cap primarily secreted by VSMCs, we measured the percentage of VSMCs in the plaque area. Remarkably, our results demonstrated a significant reduction in the area occupied by a-SMA positive VSMCs within the plaque of Mzb1-overexpressing mice compared to ApoE−/− mice (Fig. 5a, f). In addition, we calculated the vulnerability plaque index for each group, and our results indicated that the overexpression of Mzb1 substantially decreased the vulnerability index of plaques (Fig. 5g). These findings provide strong evidence that Mzb1 holds great promise for enhancing the stability of atherosclerotic plaques.

Mzb1 Attenuates Atherosclerotic Plaque Vulnerability in ApoE.−/− Mice. Aortic roots were subjected to Oil-red O staining, H&E staining, Masson staining, and immunohistochemical staining for CD68 and α-SMA in mice (a). Plaque size in the aortic root was evaluated using Oil-red O staining (b) and the size of the necrotic core of the aortic root plaque was assessed through H&E staining (c). Collagen fraction was evaluated using Masson staining (d), and immunohistochemical staining was performed for CD68 and αSMA (e, f). The vulnerability index changes in aortic root plaques (g). Scale bar: 500 μm; *p < 0.05; ***p < 0.001; ****p < 0.0001
Mzb1 Suppresses Atherosclerotic Plaque-Induced Apoptosis and Maintains the Structure and Function of Mitochondria
Additionally, we observed significantly higher levels of Bax and Cyt-c expression, and significantly lower levels of Bcl-2 expression in HFD mice than in mice in the sham group. Notably, these differences were nullified by the overexpression of Mzb1 (Fig. 6a-d), highlighting the role of Mzb1 in suppressing the expression of Bax and Cyt-c. To further investigate the effects of Mzb1, we examined the ultrastructure of the aorta using TEM. Electron photomicrographs of the aortas from the sham and Mzb1-transfected groups exhibited normal mitochondria. Conversely, in HFD-induced AS mice transfected with the vector, there was evidence of mitochondrial aggregation and enlargement. Detailed analysis of the images revealed swollen and disorganized mitochondria, along with fragmented structures indicative of impaired respiratory capacity. Importantly, overexpression of Mzb1 successfully mitigated the detrimental effects of AS, preserving the structure and function of the mitochondria (Fig. 6e). Consistent with previous studies, our findings demonstrated a downregulation in the expression of SIRT1 and the level of AMPK, an upstream component of SIRT1, in HFD mice. However, these changes were effectively restored by the overexpression of Mzb1 (Fig. 6f, g). These results strongly indicate that Mzb1 influences mitochondrial function by modulating the AMPK/SIRT1 signaling pathway.

Mzb1 suppresses atherosclerotic plaque-induced apoptosis and maintains the structure and function of mitochondria. Representative Western blot bands depicting Bax, Bcl-2 and Cyt-c in different groups (a-d). Electron microscopic findings of mitochondria ultrastructure. The red triangle indicates swollen and disorganized mitochondria and fragmented mitochondria. The yellow pentagram indicates a normal mitochondrial ultrastructure (e). The effect of Mzb1 on the protein levels of AMPK and SIRT1 in different groups (f–h). Scale bar = 2 μm, *p < 0.05; **p < 0.01; ****p < 0.0001
Discussion
In this study, we investigated the protective role of Mzb1 in attenuating atherosclerotic plaque vulnerability. We observed a significant decrease in Mzb1 expression in mouse models of AS and in HVSMCs treated with ox-LDL in vitro. The reduced Mzb1 expression correlated with increased susceptibility to plaques in vivo and heightened apoptosis of ox-LDL-treated HVSMCs in vitro. In contrast, overexpression of Mzb1 improved plaque stability and alleviated ox-LDL-induced apoptosis in HVSMCs. Furthermore, we found that Mzb1 plays a crucial role in enhancing mitochondrial function in ox-LDL-treated HVSMCs, as evidenced by the restoration of decreased mitochondrial ATP synthesis, mitochondrial membrane potential, and reduced production of mtROS. Moreover, our findings suggest that Mzb1 overexpression regulates the AMPK/SIRT1 signaling pathway, leading to the attenuation of atherosclerotic plaque vulnerability. Overall, our study provides compelling evidence that Mzb1 exerts a protective effect on atherosclerotic plaques by modulating mitochondrial function in ApoE−/− mice.
Numerous studies have consistently shown that VSMCs play a crucial role in the development of atherosclerosis, which involves thickening of the inner layer of the intima and formation of atheromatous plaques [23, 24]. The loss of VSMCs has been identified as a contributing factor to plaque instability and subsequent complications [24]. VSMC apoptosis is believed to be a major contributor to atherosclerotic plaques [25]. Importantly, studies have demonstrated that elevated levels of ox-LDL, a key player in atherogenesis, can significantly induce VSMC apoptosis [26]. Our results further underscore the significance of this process by revealing that Mzb1 treatment mitigates ox-LDL-induced VSMC apoptosis. This suggests a potential therapeutic strategy for preventing plaque instability.
Mzb1, which encodes a B-cell-specific endoplasmic reticulum (ER)-localized protein, is an important factor in immune protein synthesis. Acting as a chaperone for Grp94/gp96 (Hsp90b1) under ER stress, Mzb1 facilitates efficient antibody secretion both in vitro and in immunized mice [27, 28]. Previous studies have mainly focused on understanding the function of Mzb1 in the context of ER stress. The ER is responsible for vital cellular processes, such as protein folding and the transport of secretory and membrane proteins necessary for maintaining cellular homeostasis [29]. However, accumulation of misfolded proteins in the ER triggers the unfolded protein response (UPR), which plays a crucial role in the onset, progression, and clinical outcomes of atherosclerosis [30,31,32]. Moreover, ER stress is a key mechanism regulating plaque vulnerability and acute complications associated with atherosclerosis [33]. Previous studies have demonstrated that Mzb1 regulates the expression of GRP94 and GRP78, which are involved in ER stress and affect the classical ER stress signaling pathway [21, 28].
The UPR is a cellular mechanism activated in response to ER stress, leading to a cascade of detrimental effects, including oxidative stress, vascular cell apoptosis, and the development of atherosclerosis [32, 34]. Mitochondrial dysfunction results in an increased production of superoxides, which, in turn, causes the oxidation of low-density lipoproteins [35]. This oxidation promotes vascular cell apoptosis and increases atherosclerotic lesions [36]. Recent research has highlighted extensive communication between the ER and mitochondria, suggesting significant crosstalk between these organelles under normal physiological conditions, as well as in pathophysiological states [37]. Understanding the key players involved in this crosstalk is crucial for gaining insight into the underlying mechanisms of ER and mitochondrial dysfunction and their impact on atherosclerosis.
Mitochondrial dysfunction plays a significant role in the development of numerous chronic diseases, including atherosclerosis [38]. Reduced mitochondrial quantity has been extensively linked to the progression of atherosclerosis [39]. In the presence of atherogenic conditions, cellular increases in reactive oxygen species (ROS) have been reported to cause damage to the mitochondria, leading to further production of ROS in a continuous cycle [36]. Damage to mitochondrial DNA (mtDNA) caused by ROS has been found to correlate not only with the extent of atherosclerotic plaques but also with the early stages of lesion formation [40]. Research has demonstrated that reduced ATP production as a result of these defects is associated with apoptosis and the inhibition of cell proliferation. Apoptosis induced by ox-LDL cholesterol involves mitochondrial calcium overload, loss of mitochondrial membrane potential, and the release of cytochrome c, playing a crucial role in the initiation and progression of apoptosis [41]. Ultimately, these processes lead to plaque vulnerability [42]. Our study showed that Mzb1 increased mitochondrial ATP production and mitochondrial membrane potential in ox-LDL-induced HVSMCs. Mzb1 also significantly reduced mitochondrial ROS synthesis.
Previous human pathological examinations demonstrated that plaque components, rather than their size, play a more prominent role in the development of atherosclerotic plaques. Increased plaque vulnerability is a major cause of plaque disruption [43, 44]. Vulnerable plaques are characterized by higher lipid and macrophage content and reduced collagen and SMCs [44]. Our study revealed that atherosclerotic lesions in ApoE−/− + Mzb1-oe mice displayed a less vulnerable status compared to ApoE−/− mice. This was evidenced by a smaller necrotic core size, increased collagen content, reduced macrophage infiltration, and an elevated number of VSMCs within the lesions.
In conclusion, the findings of this study strongly indicate that Mzb1 plays a significant role in reducing the vulnerability of atherosclerotic plaques by modulating mitochondrial function and mitigating apoptosis. These results provide valuable insights into potential targets and interventions for treating impaired mitochondrial energy metabolism in atherosclerotic plaques. By understanding and targeting these mechanisms, new approaches could be developed to address the underlying causes of atherosclerosis.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- AS:
-
Atherosclerosis
- HVSMCs:
-
Human vascular smooth muscle cells
- ox-LDL:
-
Oxidized low-density lipoprotein
- ROS:
-
Reactive oxygen species
- ER:
-
Endoplasmic reticulum
- HFD:
-
High fat diet
- TEM:
-
Transmission electron microscopy
- UPR:
-
Unfolded protein response
References
Kolodgie FD, et al. The thin-cap fibroatheroma: a type of vulnerable plaque - The major precursor lesion to acute coronary syndromes. Curr Opin Cardiol. 2001;16(5):285–92.
Finn AV, et al. Concept of Vulnerable/Unstable Plaque. Arterioscler Thromb Vasc Biol. 2010;30(7):1282–92.
Bentzon JF, et al. Mechanisms of Plaque Formation and Rupture. Circ Res. 2014;114(12):1852–66.
Sukhovershin RA, et al. Local Inhibition of Macrophage and Smooth Muscle Cell Proliferation to Suppress Plaque Progression. Methodist Debakey Cardiovasc J. 2016;12(3):141–5.
Basatemur GL, et al. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. 2019;16(12):727–44.
Grootaert MOJ, Bennett MR. Vascular smooth muscle cells in atherosclerosis: time for a re-assessment. Cardiovasc Res. 2021;117(11):2326–39.
Forte M, et al. The role of mitochondrial dynamics in cardiovascular diseases. Br J Pharmacol. 2021;178(10):2060–76.
Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Investig. 2018;128(9):3716–26.
Iglewski M, et al. Mitochondrial Fission and Autophagy in the Normal and Diseased Heart. Curr Hypertens Rep. 2010;12(6):418–25.
Lee CF, et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation. 2016;134(12):883–94.
Tsai K-L et al. Chlorogenic acid protects against oxldl-induced oxidative damage and mitochondrial dysfunction by modulating sirt1 in endothelial cells. Mol Nutr Food Res. 2018;62(11):e1700928.
Salnikova D et al. Mitochondrial dysfunction in vascular wall cells and its role in atherosclerosis. Int J Mol Sci. 2021;22(16):8990.
Suarez-Rivero JM et al. From mitochondria to atherosclerosis: the inflammation path. Biomedicines. 2021;9(3):258.
Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007;100(4):460–73.
Huang Y et al. MZB1-expressing cells are essential for local immunoglobulin production in chronic rhinosinusitis with nasal polyps. Annals of allergy, asthma & immunology: official publication of the American College of Allergy, Asthma, & Immunology. 2023. https://doi.org/10.1016/j.anai.2023.10.008. Online ahead of print.
Andreani V, et al. Cochaperone Mzb1 is a key effector of Blimp1 in plasma cell differentiation and β1-integrin function. Proc Natl Acad Sci USA. 2018;115(41):E9630–9.
Li D, et al. MZB1 targeted by miR-185-5p inhibits the migration of human periodontal ligament cells through NF-κB signaling and promotes alveolar bone loss. J Periodontal Res. 2022;57(4):811–23.
Miyagawa-Hayashino A et al. Increase of MZB1 in B cells in systemic lupus erythematosus: proteomic analysis of biopsied lymph nodes. Arthritis Res Ther. 2018;20(1):13.
Watanabe M et al. MZB1 expression indicates poor prognosis in estrogen receptor-positive breast cancer. Oncol Lett. 2020;20(5):198.
Wu W et al. COL1A1 and MZB1 as the hub genes influenced the proliferation, invasion, migration and apoptosis of rectum adenocarcinoma cells by weighted correlation network analysis. Bioorg Chem. 2020;95:103457.
Zhang L, et al. Mzb1 protects against myocardial infarction injury in mice via modulating mitochondrial function and alleviating inflammation. Acta Pharmacol Sin. 2021;42(5):691–700.
Zhou H, et al. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018;25(6):1080–93.
Bennett MR, Sinha S, Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res. 2016;118(4):692–702.
Grootaert MOJ, et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res. 2018;114(4):622–34.
Shioi A, Ikari Y. Plaque Calcification During Atherosclerosis Progression and Regression. J Atheroscler Thromb. 2018;25(4):294–303.
Ding Z, et al. Regulation of autophagy and apoptosis in response to ox-LDL in vascular smooth muscle cells, and the modulatory effects of the microRNA hsa-let-7g. Int J Cardiol. 2013;168(2):1378–85.
Flach H, et al. Mzb1 Protein Regulates Calcium Homeostasis, Antibody Secretion, and Integrin Activation in Innate-like B Cells. Immunity. 2010;33(5):723–35.
Rosenbaum M, et al. MZB1 is a GRP94 cochaperone that enables proper immunoglobulin heavy chain biosynthesis upon ER stress. Genes Dev. 2014;28(11):1165–78.
Liu M-Q, Chen Z, Chen L-X. Endoplasmic reticulum stress: a novel mechanism and therapeutic target for cardiovascular diseases. Acta Pharmacol Sin. 2016;37(4):425–43.
Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med. 2010;16(4):396–9.
Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–29.
Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107(7):839–50.
Myoishi M, et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116(11):1226–33.
Margittai E, Sitia R. Oxidative protein folding in the secretory pathway and redox signaling across compartments and cells. Traffic. 2011;12(1):1–8.
Mabile L, et al. Mitochondrial function is involved in LDL oxidation mediated by human cultured endothelial cells. Arterioscler Thromb Vasc Biol. 1997;17(8):1575–82.
Peng W, et al. Mitochondrial dysfunction in atherosclerosis. DNA Cell Biol. 2019;38(7):597–606.
Walter L, Hajnóczky G. Mitochondria and endoplasmic reticulum: The lethal interorganelle cross-talk. J Bioenerg Biomembr. 2005;37(3):191–206.
Yu EPK, Bennett MR. The role of mitochondrial DNA damage in the development of atherosclerosis. Free Radical Biol Med. 2016;100:223–30.
Yu EPK, et al. Mitochondrial respiration is reduced in atherosclerosis, promoting necrotic core formation and reducing relative fibrous cap thickness. Arterioscler Thromb Vasc Biol. 2017;37(12):2322–32.
Ciccarelli G et al. Mitochondrial dysfunction: the hidden player in the pathogenesis of atherosclerosis?. Int J Mol Sci. 2023;24(2):1086.
Yu S, et al. PACS2 is required for ox-LDL-induced endothelial cell apoptosis by regulating mitochondria-associated ER membrane formation and mitochondrial Ca2+ elevation. Exp Cell Res. 2019;379(2):191–202.
Yu E, et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation. 2013;128(7):702–12.
Rohwedder I, et al. Plasma fibronectin deficiency impedes atherosclerosis progression and fibrous cap formation. EMBO Mol Med. 2012;4(7):564–76.
Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. 2015;278(5):483–93.
Funding
This research was funded by the Natural Science Foundation of Shanghai (20ZR1411800 and 22ZR1412100).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Informed Consent
No human studies were carried out by the authors for this article. All institutional and national guidelines for the care and use of laboratory animals were followed and approved by the appropriate institutional committees.
Conflicts of Interest
No potential conflict of interest was reported by the authors.
Additional information
Associate Editor Enrique Lara-Pezzi oversaw the review of this article
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhu, G., Li, Y., Gao, H. et al. Mzb1 Attenuates Atherosclerotic Plaque Vulnerability in ApoE-/- Mice by Alleviating Apoptosis and Modulating Mitochondrial Function. J. of Cardiovasc. Trans. Res. 17, 782–794 (2024). https://doi.org/10.1007/s12265-024-10483-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-024-10483-0