
Abstract
T cells, a key component of cancer immunotherapy, undergo a variety of histone modifications and DNA methylation changes since their bone marrow progenitor stages before developing into CD8+ and CD4+ T cells. These T cell types can be categorized into distinct subtypes based on their functionality and properties, such as cytotoxic T cells (Tc), helper T cells (Th), and regulatory T cells (Treg) as subtypes for CD8+ and CD4+ T cells. Among these, the CD4+ CD25+ Tregs potentially contribute to cancer development and progression by lowering T effector (Teff) cell activity under the influence of the tumor microenvironment (TME). This contributes to the development of therapeutic resistance in patients with cancer. Subsequently, these individuals become resistant to monoclonal antibody therapy as well as clinically established immunotherapies. In this review, we delineate the different epigenetic mechanisms in cancer immune response and its involvement in therapeutic resistance. Furthermore, the possibility of epi-immunotherapeutic methods based on histone deacetylase inhibitors and histone methyltransferase inhibitors are under investigation. In this review we highlight EZH2 as the principal driver of cancer cell immunoediting and an immune escape regulator. We have addressed in detail how understanding T cell epigenetic regulation might bring unique inventive strategies to overcome drug resistance and increase the efficacy of cancer immunotherapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Since its inception, the field of “epigenetics” has expanded revealing its role in population survival, human developmental programming, and immune system adaptation. Epigenetic modifications, including DNA methylation, histone posttranslational modifications, and noncoding RNAs, exert intricate control over T cell development and function [1]. They play a fundamental role in mediating the adaptive capabilities of T cells and their ability to respond to environmental cues [1]. By understanding the epigenetic mechanisms, we can gain insights into how T cells differentiate, form stable lineages, and adapt to changing conditions. This comprehensive review explores the mechanistic aspects of epigenetic regulation in T cells and highlights its significance in the fields of immunology and cancer research.
Central to immune responses, the immune cells comprise of various subsets, including T and B lymphocytes, derived from the thymus and bone marrow, respectively. Notably, T cell development involves dynamic DNA rearrangements facilitated by DNA methylation and histone modifications, which helps to orchestrate the intricate process of lineage commitment and stability. Recent investigations have significantly advanced our knowledge of T cell subsets and their developmental trajectory, shedding light on the critical role of the epigenetic regulation. Antigen stimulation guided by specific cytokines directs T cell differentiation [2]. Chromatin remodeling, a key process driven by epigenetic modifications, modulates the transcriptional accessibility of key cytokine genes such as IL-17, IFN-g, and IL-4, thereby influencing T cell activation and effector functions [2]. Based on this cytokine secretion pattern, T cells are further categorized into distinct subsets: CD8+ and CD4+ T cells which are committed to their lineage of cytotoxic T cell (Tc cells) and helper T cell (Th cells), respectively.
Within the CD4+ T cell population, natural regulatory T cells (nTregs) and natural killer T cells (NKT cells) represent unique subsets that arise from distinct differentiation processes in the thymus [3]. Tregs have emerged as sophisticated regulators of immune responses, suppressing the proliferation and effector functions of various immune-responsive cells, including NK cells, Tc cells, B cells, and antigen-presenting cells (APCs) [4]. Studies utilizing thymectomized mice and subsequent rescue experiments with CD4+ T cell transfers have provided valuable insights into the essential role of these cells in immune control and suppression [5, 6]. The development and differentiation of Tregs are intricately governed by specific transcription factors that orchestrate transcriptional programs, and ultimately suppress the expression of immune-responsive cells [7]. This intricate mode of epigenetic regulation allows the modulation functionality of Tregs in diverse pathological conditions, including cancer [7].
This comprehensive review delves into the molecular intricacies of epigenetic regulation in T cell development and function. Unravelling the underlying epigenetic mechanisms provides critical insights into immunological processes and offers potential avenues for therapeutic interventions in cancer. The elucidation of epigenetic controls governing human T cell development holds promise for the creation of new approaches to manipulate immune responses and overcome drug resistance, ultimately advancing immunotherapeutic strategies in cancer treatment and prevention.
2 T cell maturation stages and epigenetic regulation
The various types of T cells originate from progenitor cells derived from the bone marrow. These cells undergo lineage commitment, progressing through different stages and expressing distinct clusters of differentiation (CD) surface markers [8]. This maturation process, known as lymphopoiesis or lymphoid hematopoiesis, involves specific epigenetic modifications that enable the functionality of relevant genes, while repressing alternative lineage genes [9]. During lymphoid lineage differentiation, CpG methylation plays a crucial role in both thymic development and peripheral differentiation [10]. Genes associated with the lymphoid lineage get DNA hypomethylation, while myeloid lineage genes get DNA hypermethylation [10].
One of the major regulators involved in T cell differentiation is Ikaros, a transcription factor that plays a crucial role in chromatin remodeling. Ikaros targets the nucleosome remodeling and deacetylase (NuRD) chromatin remodeler complex [11,12,13]. The NuRD complex consists of various components, including HDAC1, HDAC2, MTA1, MTA2, MBD3, Rbp46/48, and Mi-2β [14]. The interaction between Ikaros and the NuRD complex helps regulate the chromatin topology and gene expression through histone deacetylase activity [11,12,13]. Previous studies have shown that Mi-2β, a component of the NuRD complex, downregulates stem cell genes (like Mdmdc2, Mpl, Ndn, Tgm2, Tek, Ebi3) and early myeloid-promoting genes (like Csf1r, Il6ra, Egr1, Il6st) [15]. In contrast, Ikaros functions in downregulating the stem cell program in lympho-myeloid primed progenitors (LMPPs) through the formation of heterochromatin region by increasing H3K9Me3 and decreasing H3K9Ac and upregulating lymphoid-promoting genes, such as Ptcra (pre-TCRα) [16, 17], Cd4 [18], and TdT (terminal deoxynucleotidyl transferase) [19] (Fig. 1A and B). Even the mice lacking Ikaros gene expression develop thymomas which lead them to attain a phenotype similar to pre-T cell which after its reintroduction instantly led to its rapid differentiation to DP (double-positive)-like thymocytes [20]. Apart from this the Chip-Seq analysis of human B-ALL cells — Nalm6 cell line (B cell acute lymphoblastic leukemia) has shown an association of Ikaros with HDAC1 thus forming Ikaros-HDAC1 complex over 934 gene targets which characterized the presence of strong H3K27Me3 and H3K4Me3 with moderate H3K9Me3 and absence of H3K9Ac [21]. Thus, after T cells are activated, Ikaros helps in recruiting Mi-2/HDAC to these heterochromatin regions, thereby restructuring the chromatin for lymphocyte differentiation [11].

Epigenetic mechanism of T cell differentiation from HSC. A Active chromatin marks like H3K4 trimethylation and H3K9 acetylation on promoter of stem cell genes like MP1, TGM2, and MDMC2 leading to stem cell regeneration. B Repressive chromatin complexes like Ikaros and NuRD complex or Ikaros and PRC2 complex inhibit the transcription of myeloid-promoting genes leading to transition of DN1- to DN3-. C Repressive chromatin complexes like IKAROS and PRC2 or IKAROS and DNMT1 inhibit the transcription of myeloid-promoting genes and removal of these complexes activates the transcription of lymphoid-promoting genes leading to transition of DN4 to DP
Interestingly, Ikaros has been found to regulate double-negative thymocytes through polycomb repressive complex 2 (PRC2)-mediated gene silencing, independent of the NuRD complex [22]. It has been shown that Ikaros binding results in recruitment of PRC2 which causes suppression of various genes like Cd9, Ctnnd1, Glt1d1, and Cttnbp2nl through H3K27 trimethylation in the DN3 stage (double negative) [22]. Interestingly Ikaros and PRC2 also have been found to downregulate the transiently elevated expression of certain Notch target genes, including Hes1, Scn4b, and Mpzl2 (Fig. 1B) in DN2 and DN3 cells [23]. It was found that it requires Ikaros to be efficiently silenced in DN4 and DP cells, although IKAROS expression decreases in the DP thymocytes, but the gene repression is still sustained through the epigenetic memory [22].
Ikaros also plays a role in regulating DNA methylation. It has been observed that Ikaros colocalizes with DNA methyltransferase at the replication fork during the S phase of the cell cycle [24]. Remarkably, deletion of the DNA methyltransferase DNMT1 during thymic development at the double-negative stage leads to a significant reduction in double-positive T cells and mature peripheral T cells [25]. Recent studies have used Comprehensive High-throughput Arrays for Relative Methylation (CHARM) to identify differentially methylated regions (DMRs) associated with gene expression datasets [26]. These studies have revealed the negative regulation of TGF-beta signalling by selective hypomethylation of SMAD7 during early stages of thymocyte development [26] (Fig. 1C). Furthermore, genes such as Arl4c and Lck undergo DNA demethylation during lymphopoiesis, while Gcnt2 transcripts are hypermethylated and downregulated, suggesting their role in aiding myeloid potential and their loss during lymphoid lineage commitment [26].
Following T cell development, the transcription factor Bcl11b plays a crucial role in suppressing self-renewal and multipotency in double-negative thymocytes [27,28,29]. Knockdown of Bcl11b leads to the inhibition of thymocyte differentiation and accumulation in the double-negative stage, accompanied by impaired T cell development [27]. Additionally, Bcl11b has been found to reprogram DN2,3 and double-positive (DP) thymocytes into natural killer (NK)-like cells, suggesting its role in determining cell fate [30].
The CD4 locus provides another example of epigenetic regulation during T cell development [31]. In DP T cells, the CD4 locus is hypermethylated, but CD4 expression remains high compared to double-negative (DN) cells and cytotoxic CD8+ T cells [32]. Enhancer sequences E4(p) and E4(m), located upstream of the CD4 transcription start site, drive optimal CD4 expression in DP-T cells, resulting in the growth of single-positive CD4+CD8− T cells [32]. Transcription factors such as TCF1 [32], LEF1 [32], Bcl11b [33], and Satb1 [34] are involved in regulating the expression of these enhancers. Following the expression of E4(p) and E4(m), DNA demethylases TET1 and 3 cause demethylation of CD4 during thymocyte maturation, which results in the formation of single-positive (SP) CD4+CD8− T cells [32] (Fig. 2A).

Epigenetic regulation of T cell maturation. A TET1/3 binds to promoter and EXON1 of CD4 removing methylation from DNA and activating its transcription, thus maturing to CD4+ CD8− CD3+ T cells. B RUNX3 binds to intronic region S4 and assists DNMT1 to hypermethylate the promoter of CD4 thereby repressing its transcription thus maturing to CD4− CD8+ CD3+ T cells
In CD8+ T cells the activity of E4m is repressed through Runx by binding at an intronic silencer S4 and thus regulating Dnmt1-mediated hypermethylation of CD4 gene [31, 35]. Once E4p and E4m are expressed it leads TET1 and TET3 to cause locus demethylation of CD4 during the thymocyte maturation which results in the formation of SP CD4−CD8+ T cells [32] (Fig. 2B).
These recent findings shed light on the intricate molecular mechanisms and epigenetic regulation involved in T cell development. Various transcription factors and protein complexes are involved in orchestrating the differentiation and lineage commitment of T cells. Further research in this field continues to uncover additional details and provide a comprehensive understanding of T cell development.
2.1 T cell subtypes
The CD4+ and CD8+ T cells are classified into several subsets based on their function and classifications. Within this group, CD4+ T cells are classified into distinct populations of Th and Treg cells, whereas CD8+ T cells are divided into numerous subsets of cytotoxic T cells.
2.2 Helper T cells
Th cells are the category of CD4+ T cells which are further categorized into Th1, Th2, and Th17 T cells. Among these, Th1 and Th2 cells play a major role in promoting B cell class switching to complement-fixing antibodies [36, 37], which provides defense against intracellular pathogens, and induces a delayed hypersensitivity response (DTH) to stop the spread of harmful bacteria [38]. Other than these, there are Th17 cells, which are essentially defined by the production of IL-17 and provide host defense against microorganisms, like extracellular bacteria and some fungi, for which Th1 or Th2 immunity is not well suited [39]. Th17 cells are crucial for host defense, but they have also drawn a lot of interest recently because they are the main pathogenesis mediators in a number of inflammatory and autoimmune diseases.
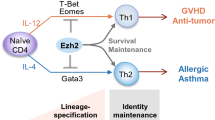
Helper T cells are subject to epigenetic regulation through repression based on H3K27Me3 on Tbx21 (Th1), Gata3 (Th2), and Rorc (Th17) [40]. This process inhibits differentiation programs that are inactive when antigenic stimulus is not present [40]. T cells undergo epigenetic reprogramming upon CD4+ activation, which involves the enrichment of the histone acetyltransferase enzyme p300 [41]. This enrichment results in the deposition of global activation marks involving histone activation and other permissive histone methylation marks, such as H3K4Me3, which enriches cytokines and their receptors, such as Il7r, Il17f, Il10, Il17a, and Il2ra [42, 43]. When these cytokines are activated, the Ifng gene in Th1 cells experiences histone acetylation due to the activation of the STAT4-T-bet axis, and the Il4 locus in Th2 experiences histone acetylation due to the activation of the STAT6-GATA3 axis [42]. Furthermore, it has been demonstrated that the engagement of the SWI/SNF chromatin remodeling complex by the Th17 master transcriptional regulator retinoic acid-related orphan receptor γt (RORγt) is necessary to promote chromatin accessibility of IL-17A, IL-17F, and IL-23R [44].
Furthermore, a subpopulation of follicular helper T cells known as Tfh cells, which express CXCR5, aid in the differentiation and maturation of B cells in response to the humoral immune response of the pathogen, which is controlled by IL-21 autocrine signalling [45]. These Tfh cell function and differentiation are determined by the BCL6 which through their signalling circuit controls the transcription factor regulating its expression [46]. In addition to an increase in expression, IL-6 signalling cascade also helps in initiating the Tfh differentiation program through the engagement of STAT1 and STAT3 promoters that in vivo increase BCL6 in CD4+ T cells [47].
2.3 T regulatory cells
Treg cells are a subset of CD4+ T cells that play an important role in immunological homeostasis and suppressing excessive immune responses. Among the CD4+ single positive (SP-CD4+) T cell population, the Treg cells, also known as thymic-derived or nTreg cells, possess distinct phenotypic characteristics [48]. They express the α chain of the IL-2 receptor (CD25) and the forkhead box P3 (FOXP3) nuclear transcription factor, which are critical for their regulatory functions [48, 49].
The development and function of Treg cells can be profoundly affected by genetic alterations at the FOXP3 locus on the chromosomes [50]. Mutations in this region can lead to dysregulation of Treg cell function, resulting in lymphoproliferative and autoimmune diseases, and in severe cases, even death [50]. To better understand the regulation of FOXP3 expression, scientists examined the alignment of the FOXP3 locus across different species. This analysis has revealed the presence of highly conserved regions, including the promoter and three non-coding regions, which collectively regulate the expression of the FOXP3 gene [51]. These noncoding regions are referred to as conserved non-coding sequences (CNS) 1, 2, and 3, which function as an enhancer facilitating the binding of various transcription factors that induce FOXP3 expression [51]. Among these conserved regions, CNS-1 is located 2 kb downstream of the transcription start site (TSS) and serves as a sensor for transforming growth factor-beta (TGF-β) [51]. CNS1 contains binding sites for SMAD proteins and NFAT, allowing it to respond to TGF-β signalling [52]. Upon stimulation of the T cell receptor (TCR) and TGF-β, there is an induction of H3K4Me3 at CNS1, which leads to the opening of the chromatin structure and allows access to Smad3 [53]. Another conserved region, CNS2, stabilizes FOXP3 expression and has been demonstrated to be crucial for FOXP3 induction. Remarkably, it has been shown that protein called Bcl11b associates with both CNS1 and CNS2 at the FOXP3 locus [54]. This association plays a critical role in FOXP3 expression, not only in nTreg cells but also in induced Tregs (iTregs) [54]. The expression of FOXP3 in response to TCR activation and TGF-β signalling is significantly reduced when Bcl11b is absent, highlighting the importance of Bcl11b in the generation of iTreg cells [54].
Apart from the genetic and epigenetic regulation of FOXP3, Treg cells exhibit hypomethylation of DNA of specific signature gene loci, such as Ikzf4, Ctla4, and Ikzf2 [53]. These signature genes are crucial for Treg cell activity because, even in the absence of FOXP3, their hypomethylation results in the expression of a gene profile exclusive to Treg cells [53]. Additionally, antigen-presenting cell (APC) maturation and the activation of STAT5 and NFAT in response to interleukin-2 (IL-2) and CD25 TCR stimulation during inflammation are essential for determining the identification of Treg cells [55]. Activated STAT5 and NFAT interact with the hypomethylated enhancer region of FOXP3 in the CNS2, counteracting the effects of pro-inflammatory cytokines and maintaining the Treg cell phenotype [56, 57]. These intricate regulatory mechanisms are instrumental in the formation of Treg cell precursors in the thymus [58]. However, they are absent in other CD4+CD8− (CD4SP) thymocytes, underscoring the unique developmental pathway of Treg cells [58]. Studies examining FOXP3 expression and its role in Treg cell development have revealed that, when compared to steady-state Treg cells, the FOXP3 gene of activated Tregs acts as a gene repressor, downregulating the expression of genes like Il2 while simultaneously upregulating the expression of CD25 and other Treg cell markers [59, 60].
In summary, the development and function of Treg cells involve a complex interplay of genetic, epigenetic, and molecular factors. Differentiation-inducing signals, chromatin landscape remodeling, FOXP3 induction, DNA demethylation, and the stable expression of FOXP3-dependent and -independent genes are all critical steps in Treg cell lineage commitment. Understanding these processes at a molecular level can provide valuable insights into immune regulation and potentially promote the creation of innovative treatment methods for autoimmune and inflammatory diseases.
2.4 CD8+ cytotoxic T cell
CTLs, sometimes referred to as CD8+ T cells, are a subset of T cells that may cytolyze target cells with either Fas or perforin/granzymes (GZM) and produce a large amount of interferon (IFN-γ). Numerous transcription factors (TFs), chemokines, and inflammatory cytokines affect the destiny of CTLs. In addition to their ability to expand IFN-γ, various inflammatory cytokines, including IL-12, IFN-α, and IFN-β, have also been shown to support the maturation, development, and endurance of cytotoxicity [61, 62]. Tbx21 encodes T-bet, a T-box TF that aids in the expression of IFN-γ, PRF, and GZMB by cytotoxic CD8+ T cells [63]. High expression of eomesodermin (Eomes) which is an another T-box transcription factor seen in memory cells may work together with Tbx21 to control the genes that CD8+ T cells transcribe for GZM, PRF, and IFN-γ [64]. Along with it, reduced cytolytic activity in CD8+ T cells and impaired IFN-γ production are also seen in Eomes+/−Tbx21−/− mice [63].
When T cells are stimulated to become effector cells, they may develop into Tc1 or Tc2 cells. When Tc1 cells are triggered by cytokines such as IL-2 and IL-12, they may destroy certain cells either directly or indirectly [65]. Cytotoxic Tc1 cells have the ability to alter the host immune response by secreting IFN-γ or TNF-α. Helper and effector immune responses are influenced by the cytokines (IL-4, IL-5, IL-10, and IL-13) released by Tc2 T cells [65].
The generation of these T cells is influenced by a number of epigenetic factors related to epigenetic regulation. One technique that may be used is DNA demethylation, which can be actively done by a number of molecules, such as glucocorticoids, MBD2, demethylase, and DNA glycosylases [66, 67]. That being said, the molecular pathways remain unknown. IFN-γ suppresses the production of the gene in naive CD8+ T lymphocytes due to methylation at numerous CpG sites in the promoter [68]. Furthermore, it has been shown that the demethylation of the interferon promoter is associated with the production of interferon in human and mouse [67]. The hypomethylation in effector CD8+ T cell promoter regions causes additional effector genes, such as PRF and GZMB, to be significantly overexpressed [69]. Activation-induced deaminase is in charge of the methylated deamination of a neighboring base, while ten-eleven translocations (TETs) are engaged in the oxidation of the methylated base during active DNA demethylation [70]. In this aspect of DNA demethylation, EZH2 found playing a key role through its interaction with TET2 to form a DNA demethylation complex [71]. Importantly, some cellular epigenetic states may be preserved by interactions between methylation and demethylation [72].
Histone hyperacetylation has also been found in CD8+ memory T cells near the promoters of GZMB, PRF1, and IFN-γ, as well as their regulators [73]. Studies indicate that CD8+ T lymphocytes produce these genes more or less in response to induced hyperacetylation or hypoacetylation of these genes [73]. Histone modification is essential for Eomes, PRF1, and GZMB expression in CD8+ T cells. In the research published by Araki et al., they have shown the relationship between the enhanced expression of Eomes, PRF, and GZMB with positively regulating memory CD8+ T cells, which was associated with their enrichment at the proximal promoter and exon 1 on histone 3 of memory CD8+ T cells [74]. Histone H3 (H3K9Ac) acetylated lysine 9 is commonly exposed to dynamic changes associated with gene expression. It is concentrated in areas that are actively regulated, including enhancer or promoter regions in subsets of CD8+ T cells [75]. Treatment with HDAC inhibitors (HDACi) like trichostatin A was found to increase the expression of naive CD8+ T cells via upregulating the H3K9Ac levels at the T cell genes [76].
Tc9, Tc17, and Tc22 subtypes have been recently identified, adding them to the subtypes that have previously been discovered. A subpopulation of CD8+ T cells known as Tc9 cells is responsible for producing IL-9. These resilient immune cells may become effectors and settle inside tumor tissues after being adopted [77]. Recent research has shown how the transcription factor IRF1 enhances the effector function of TH9 cells and affects their anticancer characteristics. Under TH9-skewing circumstances, the transcription factor STAT1 was phosphorylated by interleukin 1β (IL-1β). As a result, IRF1 was expressed, and it bound to the promoters of Il9 and Il21 to enhance the production of cytokines from TH9 cells. Furthermore, Tc9-mediated superior antitumor response showed potent anticancer effects from TH9 cells induced by IL-1β. A range of conditions have been reported for IL-17-producing CD8+ T (Tc17) cells. Since, Tc17 cells secrete the pro-inflammatory marker cytokine IL-17, it has a high degree of plasticity towards CTL in the mouse model, and exhibit the Tc2 phenotype in the mouse model related to human disease [78, 79]. This shows their contribution into both protective and pathologic immune responses in various types of both mouse and human tissues [78, 79]. Interleukin-21 (IL-21) plays important functions in the development of Th17 cells. However, in the absence of IL17 production, the development of human naive CD8+ T cells develops into Tc22 cells through IL21 [80].
3 T cell as a player in cancer
Cancer is a multifaceted and heterogeneous disease characterized by uncontrolled and abnormal cell growth. It encompasses a range of conditions that can lead to the formation of malignant tumors, which can invade surrounding tissues and metastasize to distant sites in the body. The tumor microenvironment (TME) plays a crucial role in tumor development and progression, providing a supportive niche for cancer cells to thrive [81]. Recent scientific research has shed light on the intricate interactions between the immune system and cancer cells within TME. Immuno-oncology, a rapidly evolving field, focuses on unravelling the functions of immune cells, particularly T cells, in the context of malignancy [81]. Efforts are being made to harness the immune system’s potential to attack cancer by boosting T cell activation, recruiting and maintaining T cell responses inside the tumor, and establishing an immune-responsive TME [81].
T cells have unique metabolic requirements, which can compete with cancer cells for energy resources [82]. Unlike cancer cells, T cells do not upregulate their metabolism but rather utilize increased metabolic activity to support their growth, proliferation, and survival [83]. The process of T cell activation involves a regulated transition from a state of quiescence to an active state, initiated by immunological cues [84]. Resting T cells rely on catabolism to meet their low-energy demands, deriving ATP from the oxidative breakdown of glucose [85]. However, upon activation through TCR signalling and co-stimulation mediated by CD3/CD28, T cells rapidly increase glucose uptake and undergo glycolysis, suppressing oxidative phosphorylation [86]. This metabolic shift known as the Warburg effect results in the production of lactate as a byproduct. Although glycolysis is an inefficient process for ATP production, it allows T cells to generate substrates necessary for amino acid, nucleotide, and lipid synthesis [87, 88].
In addition to metabolic regulation, Tregs within the TME play a crucial role in tumor development and immune evasion. Tregs, characterized by the expression of CD4, CD25, and FoxP3, contribute to immunosuppression by promoting tumor immune escape and angiogenic reprogramming [89, 90]. Epigenetic factors, such as the histone methyltransferase EZH2 of the polycomb repressive complex 2 (PRC2), control Treg persistence and function [91]. EZH2 regulates the development of B cells and T cells through the suppressive post-translational modification H3K27Me3 [92, 93]. Recent studies have shown that the transcription factor c-Rel activates EZH2 transcription in lymphoid cells, including B cells and T cells, highlighting its role in regulating Treg function [94].
Furthermore, TME remodeling and STING gene expression have been shown to influence intratumoral Treg levels, influencing tumor growth, metastasis, and chemoresistance through controlling various chemokines and cytokines in the TME, which is driven by Tregs [95,96,97]. Tregs contribute to tumor metabolic reprogramming via the STING/ILC2 axis, resulting in immunosuppression and a reduction in CD4+ T cell infiltration [98]. Tregs are responsible for GATA3/NOS2-related immunosuppression by blocking STING, which results in a reduction in CD4+ T cell infiltration and an increase in metastatic lung burden [98]. Increased Treg expression was associated to a low T/MDSC ratio, resulting in a tumor-promoting situation, via the limiting of CD8+CD44+CD62L− T effector cells [98]. Tregs, in particular TIM3+/LAG3+ Tregs, produce Kras-related immunosuppressive chemoresistance and have been associated to T cell dysfunction [98]. Additionally, the presence of CD8+ T cells within the TME has been associated with positive therapeutic effects in cancer treatment. CD8+ T cells can enhance the efficacy of chemotherapy by overcoming cancer cell resistance through non-immune functions [99]. For example, CD8+ T cells can modify the metabolism of cancer associated fibroblasts (CAFs) to decrease the production of glutathione (GSH), which contributes to chemoresistance [99].
Recent studies have also highlighted the significance of CD8+ T cells in cancer treatment. CD8+ T cells play a critical role in direct cytotoxic attack against cancer cells, but their functions extend beyond this role [100]. For example, they can enhance the efficacy of chemotherapy by modulating the TME [100]. MiR-424, a micro-RNA, has been identified as a negative regulator of immune regulatory proteins, including programmed death-1 ligand 1 (PD-1L), which is involved in immune checkpoint regulation [100]. Manipulating miR-424 levels can sensitize cancer cells to chemotherapy by promoting the infiltration and function of CD8+ T cells [100].
Non-immune activities of CD8+ T cells are also being investigated. As previously noted, these T cells can influence cancer cell resistance to chemotherapy by altering the metabolism of CAFs to reduce GSH synthesis [99]. In addition, CD4+ Th cells are critical in directing immune responses against cancer. Th cells regulate the activity of CD8+ T and B cells, making them crucial components of adaptive immunity.
Thus, ongoing research efforts focusing on the roles of immune cell populations, including Tregs, CD8+ T cells, and Th cells, in various cancer types contribute to our understanding of the complex dynamics within the TME [101]. These studies provide valuable insights into the intricate interplay between immune cells and the TME, offering potential targets for therapeutic interventions. Enhancing our comprehension of the dynamic interactions within the TME is vital for developing effective immunotherapies and improving outcomes for cancer patients.
4 T cell-mediated immunotherapy
Immunotherapy is a type of biological therapy in which the immune system is regulated using substances produced by living organisms, allowing our body to fight infections and other diseases such as cancer more effectively. T cell activation controls and guides this therapy, acting as a checkpoint molecule to regulate hyperactivation and fine-tune the immune response. In this context, the two most potent examples are cytotoxic T lymphocyte antigen 4 (CTLA4) and programmed cell death 1 (PD1), which complement each other’s function while ensuring self-tolerance of the T cell response regardless of their temporal and spatial expression patterns [102].
CTLA4 inhibits T cell activation by directly antagonizing CD28 through competition for costimulatory ligands and thus recruiting inhibitory effectors [103]. CTLA4 also expresses on Treg cells, where it directly regulates the immunosuppressive activity of these cells [104]. In vitro studies have also validated CTLA4’s critical role in the release of anti-inflammatory cytokines, which leads to the regulation of T cell proliferation in the vicinity [105, 106]. As a result of which, CTLA4 expressing Treg cells tries to compensate, for the absence of conventional T cells expressing CTLA4 in Rag−/− mice mediated by CTLA4 antibody-mediated neutralization [107]. It has been suggested that by inhibiting CTLA4, which has been shown to be a negative regulator of T cell activation, T cells may mount a therapeutic response against cancer [108]. Later it was validated that CTLA4-mediated tumor regressive mechanisms are pleiotropic, but are linked by the action of regulating a single cell type, the T lymphocyte.
The other mode of immunotherapeutic regulation is by PD1 that was identified as a potential mediator of apoptosis [109]. However, further studies and additional data indicated that it also functions similar to CTLA4 in controlling immune system hyperactivity [109]. PD1 suppresses immune responses primarily by inhibiting intracellular signalling in effector T and Treg cells [110]. This research led to the discovery of PD1 axis as a negative regulator for T cell activation, which was later used by several researchers for the preclinical target of cancer treatment. Using mAbs or secreted PD1 extracellular domains to block the PD1 axis could reverse the effect of CD8+ T cell cytotoxic antitumor response and increased T cell cytotoxicity against tumor cells [111,112,113]. The similar results were validated in vivo, where the PD1 inhibition was found to not only enhance the antitumor immunity but also limit the metastasis in B16 melanoma and CT26 colon carcinoma [114]. Thus, blocking PD1/PDL1 can improve tumor cytolysis while also limiting metastasis.
The separate modulation of CTLA4- and PD1-mediated immunotherapeutics has resulted in their acceptance as a treatment strategies for a wide spectrum of malignancies including cancer.
5 T cell-mediated drug resistance
Drug resistance has emerged as a formidable obstacle in the battle against cancer. The phenomenon of therapeutic resistance encompasses a complex web of independent or interconnected mechanisms, including both intrinsic and acquired chemoresistance. However, there is a pressing need to comprehend the poorly defined transient or conditional resistance mechanism, which often underlies chemotherapy failure. Notably, malignant ascites spheroids and detachable cancer cells exhibiting stem cell-like characteristics [115] stand out as prime suspects in conferring resistance. Understanding these mechanisms is crucial for developing effective strategies to overcome drug resistance and enhance cancer treatment efficacy.
The tumor immune microenvironment and the emergence of treatment resistance are intrinsically intertwined, forming a critical junction for intervention. Within the TME, cancer cells undergo constant adaptations to evade recognition by immune effector cells [116, 117]. These adaptations involve diminishing the expression of neoantigens (non-self-antigens), antigen presentation molecules such as MHC-I [118, 119], and the mutational burden. As a result, the ability of the tumor to respond to adaptive immune responses is hampered, resulting in the development of intrinsic tumor characteristics such as altered expression of immune regulatory molecules. Furthermore, tumor-extrinsic factors such as immunosuppressive cells, soluble suppressive chemicals, and inhibitory receptors on immune cells help to reshape the tumor-infiltrating lymphocyte (TIL) landscape. This remodeling is characterized by an imbalance in the ratio of T regulatory cells to T effector cells, impairing the function of T effector cells and fostering tumor growth and metastasis [120].
The immunosuppressive milieu orchestrated by cancer cells extends beyond cellular interactions, encompassing the secretion of various cytokines (e.g., interleukin-10 (IL-10)) [121] and growth factors (such as transforming growth factor-β [TGF-β] and vascular endothelial growth factor [VEGF]) [122]. Moreover, the secretion of immunosuppressive cytokine IL-10 by cancer stem cells (CSCs) expedites the differentiation of CD4+CD25+FOXP3+ Treg cells [123]. Additionally, cancer cells bolster immunosuppression and tumor progression by enhancing the recruitment and activation of immunosuppressive cells like Tregs, B regulatory cells (Bregs), tumor-associated macrophages, and myeloid-derived suppressor cells (MDSCs). They also upregulate immune checkpoint molecules, including CTLA-4 and PD-1, on immune cells [120]. These orchestrated factors collectively obstruct clinical outcomes and response rates to cancer therapy. Thus, unravelling the intricate dynamics of drug resistance and immune escape in cancer is essential for devising effective therapeutic strategies. Understanding the mechanisms behind tumor evolution, immune evasion, and the TME’s immunosuppressive nature will pave the way for improved treatment outcomes and strategies to overcome resistance.
6 Epigenetic regulation of T cell-mediated drug resistance
Epigenetic processes play a critical role in the normal development and maintenance of tissue-specific gene expression programs. These intricate mechanisms can be disrupted, leading to significant alterations in gene function and the initiation of malignant cellular transformations. One hallmark of cancer is the widespread changes observed in the epigenetic environment. It is now recognized that both genetic changes and epigenetic aberrations contribute to cancer development and progression, challenging the earlier notion of cancer solely as a hereditary disease. Recent advancements in the rapidly evolving field of cancer epigenetics have unveiled extensive reprogramming across all components of the epigenetic machinery in cancer, encompassing DNA methylation, histone modifications, and non-coding RNAs.
Emerging research suggests that epigenetic regulation plays a crucial role in the immune evasion strategies employed by tumors, known as immunoediting. This phenomenon often involves the downregulation of major histocompatibility complex class I (MHC-I) molecules or loss of antigen expression, resulting in a reduced ability to present antigens to the immune system [124]. In human breast cancer, for example, DNA methylation marks at the promoter regions silence MHC-I genes [124]. Inhibition of DNA methylation through interferon treatment alters the methylation state of MHC-I gene promoters and enhances gene expression [124]. Similarly, studies have demonstrated a correlation between the downregulation of immunogenic antigens and the evasion of adoptive cell therapy [125]. Treatment with DNA demethylating agents can restore the expression of these immunogenic antigens [125].
Immunotherapy resistance can stem from the epigenetic control exerted by the PRC on MHC-I antigen presentation [126]. One of the essential functions of PRC2 is its ability to silence the MHC-I antigen processing pathway, thereby facilitating the evasion of T cell-mediated immune responses [126]. Furthermore, studies have indicated that reduced expression of the CD8 T lymphocytes has impaired antigen processing and presentation system in colon and ovarian cancer cells. Notably, treatment with DNMT inhibitors has been shown to enhance the expression of antigen processing and presentation genes, as well as cancer-testis antigens, potentially rendering patients more susceptible to immunotherapy [127].
Further investigations have demonstrated the upregulation of antigen processing and presentation machinery in prostate and breast cancer cells following treatment with HDACi [128]. This upregulation enhances the sensitivity of tumor cells to T cell-mediated lysis, indicating a loss of their ability to resist immunological attack [128]. Recent studies have highlighted the role of macroH2A1-dependent CSC-like secretome in the absence of certain immune mediators in the growth of CD4+CD25+/FoxP3+ Treg cells while suppressing CD4+CD25−/FoxP3+ T cells in human T lymphocyte populations. The formation of Tregs is traditionally dependent on IL-2 [129].
7 Epi-immunotherapy in cancer
Epi-immunotherapy has emerged as a key targeted therapeutic technique in cancer treatment in recent years. As immune evasion becomes a key barrier in restoring tumor immune recognition, many types of HDACi have proven great efficacy in suppressing tumor development [130]. Their use in conjunction with various chemotherapeutic agents and immunotherapy techniques has resulted in considerable clinical improvements for the patient [130]. However, the specific targets and mechanisms of these HDACi are not well elucidated.
The immune checkpoint factors CTLA-4 and PD-1/PD-L1 have been identified as critical targets for monoclonal antibody development in cancer immunotherapy. These MAbs have shown promising results by modulating immune inhibitory pathways. However, to overcome its limitations and ensure patient safety, there is growing evidence that combining epigenetic modulators with immunotherapy can be beneficial. In this context, EZH2, a key epigenetic regulator, has been implicated in controlling both modes of immune therapeutic regulation [131]. A detailed molecular mechanism of this regulation is discussed below.
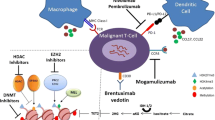
Recent findings have demonstrated that c-Rel, a transcription factor targeted for immunotherapy, plays a crucial role in melanoma growth. Chemical inhibition of c-Rel delays melanoma growth by mitigating Treg-mediated immune suppression and enhancing the response to anti-PD-1 immunotherapy [131]. In hepatoma cells, high levels of EZH2 inhibited the expression of CD274, a transcription factor responsible for PD-L1 [132]. This inhibition results in low PD-L1 expression that confers resistance against anti-PD-L1 therapy. In addition, it is shown that suppressing EZH2 expression can induce pro-inflammatory functions in tumor-infiltrating Tregs (TI-Tregs) [133]. By remodeling the TME, EZH2 suppression promotes the recruitment and activity of CD8+ and CD4+ effector T cells [133]. This leads to enhanced anticancer immunity through increased secretion of Th1-type chemokines CXCL9 and CXCL10 [133] (Fig. 3A).

Mechanistic insights into epi-immunotherapy in cancer. A Activation of chromatin repressor EZH2 by NFκB/CREL complex which leads to transcriptional repression of chemokines by chromatin repressor EZH2. B Repression of EZH2 in tumor cells using anti-PDL1 antibody leads to release of chemokines which promotes anti-CTLA4 antibody efficacy
Beyond its role in combination with anti-PD-1 therapy, the implications of EZH2 modulation have also been explored in conjunction with anti-CTLA-4 therapy [134]. Treatment with anti-CTLA-4 (Ipilimumab) upregulates EZH2 expression as a compensatory mechanism [134]. However, suppressing EZH2 using the drug CPI-1205 alongside Ipilimumab improves the response to anti-CTLA-4 therapy [134] (Fig. 3B). This combination alters the TME, modulates effector cytotoxic T cells, and transforms Tregs into effector T cells. EZH2 activation is associated with the maintenance of Treg cells by suppressing genes involved in Treg development and stabilizing their identity [135]. The increased expression of Tregs due to EZH2 activation in cancer patient was previously reported [136]. Given its role in Treg-mediated suppression, EZH2 is a prime target for epi-immunotherapy [136]. This therapeutic approach is being investigated and validated in various clinical trials utilizing different drugs to suppress EZH2 expression for the treatment of diverse cancer types. Detailed information on these trials can be found in Table 1, emphasizing the critical regulatory function of EZH2 as a target for cancer therapy.
8 Accessory mechanism of EZH2 in chemoresistance
EZH2 is involved in various mechanisms of chemoresistance in different cancer types. For instance, in glioblastoma (GBM), EZH2 contributes to temozolomide resistance by stabilizing H3K27Me3 in the promoter region of the FADD gene (FAS-associated death domain), resulting in the stabilization of PARP1 and enhanced DNA repair capacity. This resistance mechanism is mediated by ATRX (alterations of alpha thalassemia/mental retardation syndrome X-linked). In contrast, EZH2 downregulation by the lncRNA taurine-upregulated gene 1 (TUG1) sensitizes glioma cells to temozolomide (TMZ). TUG1 downregulates EZH2 expression, inhibiting cell proliferation, stem cell-like properties, and promoting apoptosis, thereby enhancing TMZ sensitivity [137].
In breast cancer, EZH2 activation leads to therapy-induced resistance by upregulating the transcription factor STAT3, which in turn increases the expression of micro-RNAs such as miR-378a-3p and miR-378d. These micro-RNAs are exosome-secreted and affect several signalling pathways, including WNT/-catenin and Notch, by targeting WNT antagonist DKK3 and Notch suppressor NUM. This process promotes the expansion of breast cancer tumor-initiating cells. Interestingly, EZH2 has a negative correlation with taxol resistance in breast cancer. It induces the deposition of H3K27Me3 on the promoter of the anti-apoptotic gene p21, resulting in its repression. This repression promotes apoptosis and enhances sensitivity to taxol [138].
EZH2 also plays a role in oxaliplatin resistance in colorectal carcinoma (CRC). In this context, the upregulated P53-inhibiting lncRNA (PiHL) interacts with EZH2 and inhibits its binding to the HMGA2 promoter. This leads to the upregulation of HMGA2 transcription, subsequent activation of the PI3K/Akt pathway, increased cell viability, and resistance to oxaliplatin treatment. Remarkably, in CRC, EZH2 promotes chemoresistance to cisplatin by upregulating the type II Na/Pi co-transporter encoded by SLC34A2. This upregulation occurs through the direct binding of HIF1A, which is dependent on reactive oxygen species production. Increased EZH2 expression downregulates apoptosis, leading to resistance against cisplatin [137].
Furthermore, EZH2 has implications in small-cell lung carcinoma (SCLC) where it is involved in gene silencing of SLFN11 by depositing H3K27Me3 at the gene body of SLFN11. This results in a decrease in SLFN11 expression, global increase in H3K27Me3 levels, and decreased H3K27Ac. Pharmacological inhibition of EZH2 in SCLC cells leads to increased SLFN11 expression and enhanced sensitivity to IR irradiation [139]. Notably, EZH2 is also implicated in acquired chemoresistance in lung adenocarcinoma. The transcription factor HOXB13, upregulated in cisplatin-resistant lung adenocarcinoma, targets the ABC transporter ABCG1 and induces its expression, conferring chemoresistance. Additionally, HOXB13 targets the EZH2 promoter, leading to EZH2 upregulation, further promoting cisplatin resistance [140].
Bladder cancer stem cells (BCSCs) also exhibit EZH2-mediated chemoresistance. EZH2 forms a complex with heterogeneous nuclear ribonucleoprotein K (hnRNPK) and low expressed in bladder cancer stem cells (lnc-LBCS) to repress the transcription factor SOX2. SOX2 is crucial for BCSC self-renewal and chemoresistance. By inhibiting SOX2 transcription, EZH2 suppresses BCSC maintenance and promotes sensitivity to gemcitabine and cisplatin [141].
In gastric cancer, EZH2 is involved in chemoresistance against 5-fluorouracil (5FU) and oxaliplatin. EZH2 inhibits the expression of FBOXO32, an F-box protein that is part of the ubiquitin protein ligase complex. EZH2 is recruited to the promoter region of FBOXO32, leading to epigenetic silencing by depositing H3K27Me3 and subsequent inhibition of apoptosis, thereby promoting chemoresistance [142]. EZH2 also antagonizes oxaliplatin resistance in gastric cancer by binding to the promoter of carbohydrate sulfotransferase 7 (CHST7), which leads to the silencing of CHST7 through H3K27Me3 deposition, thereby reversing oxaliplatin resistance [143].
EZH2 is implicated in chemoresistance in hepatocellular carcinoma. It causes deposition of H3K27Me3 on the promoter of miR-381, resulting in upregulation of SETDB1, which promotes cancer progression and confers resistance to cisplatin [144].
In multiple myeloma, EZH2 is involved in bortezomib resistance. The long non-coding RNA ANRIL, which is overexpressed in multiple myeloma cells, interacts with EZH2 and suppresses the tumor suppressor gene PTEN through H3K27Me3 deposition on its promoter. This inhibition of PTEN promotes chemoresistance against bortezomib [145].
These studies elucidate the diverse mechanisms by which EZH2 contributes to chemoresistance in various cancer types, highlighting its potential as a therapeutic target for overcoming treatment resistance.
9 Concluding remarks
The present review establishes the pivotal role of T cells in the regulation of immune responses against cancer. DNA methylation and histone modifications play a crucial role in the differentiation of T cells and the induction of a cellular adaptive immune response. The intricate process of T cell development, guided by DNMTs, Ikaros, and NuRD and PRC2 complexes, determines the fate and function of T cells within the immune system. Understanding how T cells and the immune system impact cancer initiation, progression, and therapy resistance has been a challenging aspect of immunology. For over a century, the debate regarding the immune system’s control over cancer has persisted. However, recent advancements in our understanding of the immune system and the identification of tumor antigens have shed light on the crucial role of T cells in cancer immunity. Epigenetic factors exert significant influence on the immune response to cancer, shaping T cell differentiation and function. Preclinical studies have demonstrated the potential of targeting epigenetic regulators, such as EZH2, to enhance tumor immunogenicity, improve immune cell activities, and modulate the immunosuppressive TME. Combinatorial approaches involving epi-drugs targeting EZH2 with immunotherapy have shown promise in increasing treatment efficacy. These findings highlight the importance of T cells as key players in cancer immune responses and underscore the potential of harnessing epigenetic regulation to enhance T cell-mediated anti-cancer immunity.
10 Future perspectives
Despite significant progress in understanding the functional role of epigenetics in cancer, there is still much to unravel about the complex interplay between epigenetic factors and immune control, particularly within the context of T cell responses. Given the prominent function of T cells in coordinating anti-cancer immune responses, this gives an ideal opportunity to re-evaluate current approaches to combating cancer by using immune-based therapeutics. Epigenetic alterations have been shown to globally reshape chromatin structure and dysregulate critical pathways within the TME, impacting T cell function. The emerging field of epi-immunotherapy, integrating epigenetic and immune therapies, holds tremendous potential for targeted interventions and the elimination of the immunosuppressive TME. This novel therapeutic approach not only offers renewed hope but also paves the way for innovative treatment modalities. Further exploration of the molecular mechanisms underlying epigenetic regulation in cancer cells, particularly within the context of T cell responses, will be pivotal in refining and developing effective therapeutic strategies. By deciphering the intricate molecular landscape, we can harness the power of T cells and epigenetic modulation to reset the cancer state and establish robust control over the disease.
Data availability
N/A.
References
Mostoslavsky, R., Alt, F. W., & Bassing, C. H. (2003). Chromatin dynamics and locus accessibility in the immune system. Nature Immunology, 4(7), 603–606. https://doi.org/10.1038/ni0703-603
Sawalha, A. H. (2008). Epigenetics and T-cell immunity. Autoimmunity, 41(4), 245–252. https://doi.org/10.1080/08916930802024145
Hongo, D., Tang, X., Dutt, S., Nador, R. G., & Strober, S. (2012). Interactions between NKT cells and Tregs are required for tolerance to combined bone marrow and organ transplants. Blood, 119(6), 1581–1589. https://doi.org/10.1182/blood-2011-08-371948
Takahashi, T., & Sakaguchi, S. (2003). Naturally arising CD25+CD4+ regulatory T cells in maintaining immunologic self-tolerance and preventing autoimmune disease. Current Molecular Medicine, 3(8), 693–706. https://doi.org/10.2174/1566524033479429
Sakaguchi, S., Fukuma, K., Kuribayashi, K., & Masuda, T. (1985). Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. The Journal of Experimental Medicine, 161(1), 72–87. https://doi.org/10.1084/jem.161.1.72
Sugihara, S., Izumi, Y., Yoshioka, T., Yagi, H., Tsujimura, T., Tarutani, O., Kohno, Y., Murakami, S., Hamaoka, T., & Fujiwara, H. (1988). Autoimmune thyroiditis induced in mice depleted of particular T cell subsets. I. Requirement of Lyt-1 dull L3T4 bright normal T cells for the induction of thyroiditis. The Journal of Immunology, 141(1), 105–113. https://www.ncbi.nlm.nih.gov/pubmed/2967864
Huehn, J., Polansky, J. K., & Hamann, A. (2009). Epigenetic control of FOXP3 expression: The key to a stable regulatory T-cell lineage? Nature Reviews Immunology, 9(2), 83–89. https://doi.org/10.1038/nri2474
Koch, U., & Radtke, F. (2011). Mechanisms of T cell development and transformation. Annual Review of Cell and Developmental Biology, 27, 539–562. https://doi.org/10.1146/annurev-cellbio-092910-154008
Rothenberg, E. V., Moore, J. E., & Yui, M. A. (2008). Launching the T-cell-lineage developmental programme. Nature Reviews Immunology, 8(1), 9–21. https://doi.org/10.1038/nri2232
Farlik, M., Halbritter, F., Muller, F., Choudry, F. A., Ebert, P., Klughammer, J., Farrow, S., Santoro, A., Ciaurro, V., Mathur, A., Uppal, R., Stunnenberg, H. G., Ouwehand, W. H., Laurenti, E., Lengauer, T., Frontini, M., & Bock, C. (2016). DNA methylation dynamics of human hematopoietic stem cell differentiation. Cell Stem Cell, 19(6), 808–822. https://doi.org/10.1016/j.stem.2016.10.019
Kim, J., Sif, S., Jones, B., Jackson, A., Koipally, J., Heller, E., Winandy, S., Viel, A., Sawyer, A., Ikeda, T., Kingston, R., & Georgopoulos, K. (1999). Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity, 10(3), 345–355. https://doi.org/10.1016/s1074-7613(00)80034-5
O’Neill, D. W., Schoetz, S. S., Lopez, R. A., Castle, M., Rabinowitz, L., Shor, E., Krawchuk, D., Goll, M. G., Renz, M., Seelig, H. P., Han, S., Seong, R. H., Park, S. D., Agalioti, T., Munshi, N., Thanos, D., Erdjument-Bromage, H., Tempst, P., & Bank, A. (2000). An ikaros-containing chromatin-remodeling complex in adult-type erythroid cells. Molecular and Cellular Biology, 20(20), 7572–7582. https://doi.org/10.1128/MCB.20.20.7572-7582.2000
Sridharan, R., & Smale, S. T. (2007). Predominant interaction of both Ikaros and Helios with the NuRD complex in immature thymocytes. Journal of Biological Chemistry, 282(41), 30227–30238. https://doi.org/10.1074/jbc.M702541200
Zhang, Y., Ng, H. H., Erdjument-Bromage, H., Tempst, P., Bird, A., & Reinberg, D. (1999). Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes & Development, 13(15), 1924–1935. https://doi.org/10.1101/gad.13.15.1924
Yoshida, T., Hazan, I., Zhang, J., Ng, S. Y., Naito, T., Snippert, H. J., Heller, E. J., Qi, X., Lawton, L. N., Williams, C. J., & Georgopoulos, K. (2008). The role of the chromatin remodeler Mi-2beta in hematopoietic stem cell self-renewal and multilineage differentiation. Genes & Development, 22(9), 1174–1189. https://doi.org/10.1101/gad.1642808
Bellavia, D., Mecarozzi, M., Campese, A. F., Grazioli, P., Talora, C., Frati, L., Gulino, A., & Screpanti, I. (2007). Notch3 and the Notch3-upregulated RNA-binding protein HuD regulate Ikaros alternative splicing. EMBO Journal, 26(6), 1670–1680. https://doi.org/10.1038/sj.emboj.7601626
Collins, B., Clambey, E. T., Scott-Browne, J., White, J., Marrack, P., Hagman, J., & Kappler, J. W. (2013). Ikaros promotes rearrangement of TCR alpha genes in an Ikaros null thymoma cell line. European Journal of Immunology, 43(2), 521–532. https://doi.org/10.1002/eji.201242757
Naito, T., Gomez-Del Arco, P., Williams, C. J., & Georgopoulos, K. (2007). Antagonistic interactions between Ikaros and the chromatin remodeler Mi-2beta determine silencer activity and Cd4 gene expression. Immunity, 27(5), 723–734. https://doi.org/10.1016/j.immuni.2007.09.008
Trinh, L. A., Ferrini, R., Cobb, B. S., Weinmann, A. S., Hahm, K., Ernst, P., Garraway, I. P., Merkenschlager, M., & Smale, S. T. (2001). Down-regulation of TDT transcription in CD4(+)CD8(+) thymocytes by Ikaros proteins in direct competition with an Ets activator. Genes & Development, 15(14), 1817–1832. https://doi.org/10.1101/gad.905601
Kathrein, K. L., Lorenz, R., Innes, A. M., Griffiths, E., & Winandy, S. (2005). Ikaros induces quiescence and T-cell differentiation in a leukemia cell line. Molecular and Cellular Biology, 25(5), 1645–1654. https://doi.org/10.1128/MCB.25.5.1645-1654.2005
Song, C., Pan, X., Ge, Z., Gowda, C., Ding, Y., Li, H., Li, Z., Yochum, G., Muschen, M., Li, Q., Payne, K. J., & Dovat, S. (2016). Epigenetic regulation of gene expression by Ikaros, HDAC1 and Casein Kinase II in leukemia. Leukemia, 30(6), 1436–1440. https://doi.org/10.1038/leu.2015.331
Oravecz, A., Apostolov, A., Polak, K., Jost, B., Le Gras, S., Chan, S., & Kastner, P. (2015). Ikaros mediates gene silencing in T cells through Polycomb repressive complex 2. Nature Communications, 6, 8823. https://doi.org/10.1038/ncomms9823
Geimer Le Lay, A. S., Oravecz, A., Mastio, J., Jung, C., Marchal, P., Ebel, C., Dembele, D., Jost, B., Le Gras, S., Thibault, C., Borggrefe, T., Kastner, P., & Chan, S. (2014). The tumor suppressor Ikaros shapes the repertoire of notch target genes in T cells. Science Signaling, 7(317), ra28. https://doi.org/10.1126/scisignal.2004545
Avitahl, N., Winandy, S., Friedrich, C., Jones, B., Ge, Y., & Georgopoulos, K. (1999). Ikaros sets thresholds for T cell activation and regulates chromosome propagation. Immunity, 10(3), 333–343. https://doi.org/10.1016/s1074-7613(00)80033-3
Lee, P. P., Fitzpatrick, D. R., Beard, C., Jessup, H. K., Lehar, S., Makar, K. W., Perez-Melgosa, M., Sweetser, M. T., Schlissel, M. S., Nguyen, S., Cherry, S. R., Tsai, J. H., Tucker, S. M., Weaver, W. M., Kelso, A., Jaenisch, R., & Wilson, C. B. (2001). A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity, 15(5), 763–774. https://doi.org/10.1016/s1074-7613(01)00227-8
Ji, H., Ehrlich, L. I., Seita, J., Murakami, P., Doi, A., Lindau, P., Lee, H., Aryee, M. J., Irizarry, R. A., Kim, K., Rossi, D. J., Inlay, M. A., Serwold, T., Karsunky, H., Ho, L., Daley, G. Q., Weissman, I. L., & Feinberg, A. P. (2010). Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature, 467(7313), 338–342. https://doi.org/10.1038/nature09367
Li, L., Leid, M., & Rothenberg, E. V. (2010). An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science, 329(5987), 89–93. https://doi.org/10.1126/science.1188989
Li, L., Zhang, J. A., Dose, M., Kueh, H. Y., Mosadeghi, R., Gounari, F., & Rothenberg, E. V. (2013). A far downstream enhancer for murine Bcl11b controls its T-cell specific expression. Blood, 122(6), 902–911. https://doi.org/10.1182/blood-2012-08-447839
Tydell, C. C., David-Fung, E. S., Moore, J. E., Rowen, L., Taghon, T., & Rothenberg, E. V. (2007). Molecular dissection of prethymic progenitor entry into the T lymphocyte developmental pathway. The Journal of Immunology, 179(1), 421–438. https://doi.org/10.4049/jimmunol.179.1.421
Li, P., Burke, S., Wang, J., Chen, X., Ortiz, M., Lee, S. C., Lu, D., Campos, L., Goulding, D., Ng, B. L., Dougan, G., Huntly, B., Gottgens, B., Jenkins, N. A., Copeland, N. G., Colucci, F., & Liu, P. (2010). Reprogramming of T cells to natural killer-like cells upon Bcl11b deletion. Science, 329(5987), 85–89. https://doi.org/10.1126/science.1188063
Sellars, M., Huh, J. R., Day, K., Issuree, P. D., Galan, C., Gobeil, S., Absher, D., Green, M. R., & Littman, D. R. (2015). Regulation of DNA methylation dictates Cd4 expression during the development of helper and cytotoxic T cell lineages. Nature Immunology, 16(7), 746–754. https://doi.org/10.1038/ni.3198
Issuree, P. D., Day, K., Au, C., Raviram, R., Zappile, P., Skok, J. A., Xue, H. H., Myers, R. M., & Littman, D. R. (2018). Stage-specific epigenetic regulation of CD4 expression by coordinated enhancer elements during T cell development. Nature Communications, 9(1), 3594. https://doi.org/10.1038/s41467-018-05834-w
Kojo, S., Tanaka, H., Endo, T. A., Muroi, S., Liu, Y., Seo, W., Tenno, M., Kakugawa, K., Naoe, Y., Nair, K., Moro, K., Katsuragi, Y., Kanai, A., Inaba, T., Egawa, T., Venkatesh, B., Minoda, A., Kominami, R., & Taniuchi, I. (2017). Priming of lineage-specifying genes by Bcl11b is required for lineage choice in post-selection thymocytes. Nature Communications, 8(1), 702. https://doi.org/10.1038/s41467-017-00768-1
Kakugawa, K., Kojo, S., Tanaka, H., Seo, W., Endo, T. A., Kitagawa, Y., Muroi, S., Tenno, M., Yasmin, N., Kohwi, Y., Sakaguchi, S., Kowhi-Shigematsu, T., & Taniuchi, I. (2017). Essential roles of SATB1 in specifying T lymphocyte subsets. Cell Reports, 19(6), 1176–1188. https://doi.org/10.1016/j.celrep.2017.04.038
Kojo, S., Yasmin, N., Muroi, S., Tenno, M., & Taniuchi, I. (2018). Runx-dependent and silencer-independent repression of a maturation enhancer in the Cd4 gene. Nature Communications, 9(1), 3593. https://doi.org/10.1038/s41467-018-05803-3
Finkelman, F. D., Holmes, J., Katona, I. M., Urban, J. F., Jr., Beckmann, M. P., Park, L. S., Schooley, K. A., Coffman, R. L., Mosmann, T. R., & Paul, W. E. (1990). Lymphokine control of in vivo immunoglobulin isotype selection. Annual Review of Immunology, 8, 303–333. https://doi.org/10.1146/annurev.iy.08.040190.001511
Stevens, T. L., Bossie, A., Sanders, V. M., Fernandez-Botran, R., Coffman, R. L., Mosmann, T. R., & Vitetta, E. S. (1988). Regulation of antibody isotype secretion by subsets of antigen-specific helper T cells. Nature, 334(6179), 255–258. https://doi.org/10.1038/334255a0
Cher, D. J., & Mosmann, T. R. (1987). Two types of murine helper T cell clone. II. Delayed-type hypersensitivity is mediated by TH1 clones. Journal of Immunology, 138(11), 3688–3694. https://www.ncbi.nlm.nih.gov/pubmed/2953788
Park, H., Li, Z., Yang, X. O., Chang, S. H., Nurieva, R., Wang, Y. H., Wang, Y., Hood, L., Zhu, Z., Tian, Q., & Dong, C. (2005). A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nature Immunology, 6(11), 1133–1141. https://doi.org/10.1038/ni1261
Zhu, J., Yamane, H., & Paul, W. E. (2010). Differentiation of effector CD4 T cell populations (*). Annual Review of Immunology, 28, 445–489. https://doi.org/10.1146/annurev-immunol-030409-101212
Vahedi, G., Kanno, Y., Furumoto, Y., Jiang, K., Parker, S. C., Erdos, M. R., Davis, S. R., Roychoudhuri, R., Restifo, N. P., Gadina, M., Tang, Z., Ruan, Y., Collins, F. S., Sartorelli, V., & O’Shea, J. J. (2015). Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature, 520(7548), 558–562. https://doi.org/10.1038/nature14154
Avni, O., Lee, D., Macian, F., Szabo, S. J., Glimcher, L. H., & Rao, A. (2002). T(H) cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nature Immunology, 3(7), 643–651. https://doi.org/10.1038/ni808
Fields, P. E., Kim, S. T., & Flavell, R. A. (2002). Cutting edge: Changes in histone acetylation at the IL-4 and IFN-gamma loci accompany Th1/Th2 differentiation. The Journal of Immunology, 169(2), 647–650. https://doi.org/10.4049/jimmunol.169.2.647
Lee, S., Kim, J., Min, H., & Seong, R. H. (2020). RORgammat-driven T(H)17 cell differentiation requires epigenetic control by the Swi/Snf chromatin remodeling complex. iScience, 23(5), 101106. https://doi.org/10.1016/j.isci.2020.101106
Olatunde, A. C., Hale, J. S., & Lamb, T. J. (2021). Cytokine-skewed Tfh cells: Functional consequences for B cell help. Trends in Immunology, 42(6), 536–550. https://doi.org/10.1016/j.it.2021.04.006
Jeon, Y. H., & Choi, Y. S. (2016). Follicular helper T (Tfh) cells in autoimmune diseases and allograft rejection. Immune Network, 16(4), 219–232. https://doi.org/10.4110/in.2016.16.4.219
Choi, Y. S., Yang, J. A., & Crotty, S. (2013). Dynamic regulation of Bcl6 in follicular helper CD4 T (Tfh) cells. Current Opinion in Immunology, 25(3), 366–372. https://doi.org/10.1016/j.coi.2013.04.003
Hori, S., Nomura, T., & Sakaguchi, S. (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science, 299(5609), 1057–1061. https://doi.org/10.1126/science.1079490
Trenado, A., Charlotte, F., Fisson, S., Yagello, M., Klatzmann, D., Salomon, B. L., & Cohen, J. L. (2003). Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. The Journal of Clinical Investigation, 112(11), 1688–1696. https://doi.org/10.1172/JCI17702
Bennett, C. L., Christie, J., Ramsdell, F., Brunkow, M. E., Ferguson, P. J., Whitesell, L., Kelly, T. E., Saulsbury, F. T., Chance, P. F., & Ochs, H. D. (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nature Genetics, 27(1), 20–21. https://doi.org/10.1038/83713
Zheng, Y., Josefowicz, S., Chaudhry, A., Peng, X. P., Forbush, K., & Rudensky, A. Y. (2010). Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature, 463(7282), 808–812. https://doi.org/10.1038/nature08750
Tone, Y., Furuuchi, K., Kojima, Y., Tykocinski, M. L., Greene, M. I., & Tone, M. (2008). Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nature Immunology, 9(2), 194–202. https://doi.org/10.1038/ni1549
Ohkura, N., Hamaguchi, M., Morikawa, H., Sugimura, K., Tanaka, A., Ito, Y., Osaki, M., Tanaka, Y., Yamashita, R., Nakano, N., Huehn, J., Fehling, H. J., Sparwasser, T., Nakai, K., & Sakaguchi, S. (2012). T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity, 37(5), 785–799. https://doi.org/10.1016/j.immuni.2012.09.010
Vanvalkenburgh, J., Albu, D. I., Bapanpally, C., Casanova, S., Califano, D., Jones, D. M., Ignatowicz, L., Kawamoto, S., Fagarasan, S., Jenkins, N. A., Copeland, N. G., Liu, P., & Avram, D. (2011). Critical role of Bcl11b in suppressor function of T regulatory cells and prevention of inflammatory bowel disease. Journal of Experimental Medicine, 208(10), 2069–2081. https://doi.org/10.1084/jem.20102683
Reis e Sousa, C. (2006). Dendritic cells in a mature age. Nature Reviews Immunology, 6(6), 476–483. https://doi.org/10.1038/nri1845
Feng, Y., Arvey, A., Chinen, T., van der Veeken, J., Gasteiger, G., & Rudensky, A. Y. (2014). Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell, 158(4), 749–763. https://doi.org/10.1016/j.cell.2014.07.031
Li, X., Liang, Y., LeBlanc, M., Benner, C., & Zheng, Y. (2014). Function of a Foxp3 cis-element in protecting regulatory T cell identity. Cell, 158(4), 734–748. https://doi.org/10.1016/j.cell.2014.07.030
Toker, A., Engelbert, D., Garg, G., Polansky, J. K., Floess, S., Miyao, T., Baron, U., Duber, S., Geffers, R., Giehr, P., Schallenberg, S., Kretschmer, K., Olek, S., Walter, J., Weiss, S., Hori, S., Hamann, A., & Huehn, J. (2013). Active demethylation of the Foxp3 locus leads to the generation of stable regulatory T cells within the thymus. The Journal of Immunology, 190(7), 3180–3188. https://doi.org/10.4049/jimmunol.1203473
Liu, Q., Du, F., Huang, W., Ding, X., Wang, Z., Yan, F., & Wu, Z. (2019). Epigenetic control of Foxp3 in intratumoral T-cells regulates growth of hepatocellular carcinoma. Aging (Albany NY), 11(8), 2343–2351. https://doi.org/10.18632/aging.101918
Morikawa, H., Ohkura, N., Vandenbon, A., Itoh, M., Nagao-Sato, S., Kawaji, H., Lassmann, T., Carninci, P., Hayashizaki, Y., Forrest, A. R., Standley, D. M., Date, H., Sakaguchi, S., & Consortium, F. (2014). Differential roles of epigenetic changes and Foxp3 expression in regulatory T cell-specific transcriptional regulation. Proceedings of the National Academy of Sciences of the United States of America, 111(14), 5289-5294.https://doi.org/10.1073/pnas.1312717110
Cox, M. A., Harrington, L. E., & Zajac, A. J. (2011). Cytokines and the inception of CD8 T cell responses. Trends in Immunology, 32(4), 180–186. https://doi.org/10.1016/j.it.2011.01.004
Haring, J. S., Badovinac, V. P., & Harty, J. T. (2006). Inflaming the CD8+ T cell response. Immunity, 25(1), 19–29. https://doi.org/10.1016/j.immuni.2006.07.001
Intlekofer, A. M., Takemoto, N., Wherry, E. J., Longworth, S. A., Northrup, J. T., Palanivel, V. R., Mullen, A. C., Gasink, C. R., Kaech, S. M., Miller, J. D., Gapin, L., Ryan, K., Russ, A. P., Lindsten, T., Orange, J. S., Goldrath, A. W., Ahmed, R., & Reiner, S. L. (2005). Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nature Immunology, 6(12), 1236–1244. https://doi.org/10.1038/ni1268
Pearce, E. L., Mullen, A. C., Martins, G. A., Krawczyk, C. M., Hutchins, A. S., Zediak, V. P., Banica, M., DiCioccio, C. B., Gross, D. A., Mao, C. A., Shen, H., Cereb, N., Yang, S. Y., Lindsten, T., Rossant, J., Hunter, C. A., & Reiner, S. L. (2003). Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science, 302(5647), 1041–1043. https://doi.org/10.1126/science.1090148
Kemp, R. A., Backstrom, B. T., & Ronchese, F. (2005). The phenotype of type 1 and type 2 CD8+ T cells activated in vitro is affected by culture conditions and correlates with effector activity. Immunology, 115(3), 315–324. https://doi.org/10.1111/j.1365-2567.2005.02168.x
Detich, N., Theberge, J., & Szyf, M. (2002). Promoter-specific activation and demethylation by MBD2/demethylase. Journal of Biological Chemistry, 277(39), 35791–35794. https://doi.org/10.1074/jbc.C200408200
Fitzpatrick, D. R., & Wilson, C. B. (2003). Methylation and demethylation in the regulation of genes, cells, and responses in the immune system. Clinical Immunology, 109(1), 37–45. https://doi.org/10.1016/s1521-6616(03)00205-5
Kersh, E. N., Fitzpatrick, D. R., Murali-Krishna, K., Shires, J., Speck, S. H., Boss, J. M., & Ahmed, R. (2006). Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells. The Journal of Immunology, 176(7), 4083–4093. https://doi.org/10.4049/jimmunol.176.7.4083
Scharer, C. D., Barwick, B. G., Youngblood, B. A., Ahmed, R., & Boss, J. M. (2013). Global DNA methylation remodeling accompanies CD8 T cell effector function. The Journal of Immunology, 191(6), 3419–3429. https://doi.org/10.4049/jimmunol.1301395
Bochtler, M., Kolano, A., & Xu, G. L. (2017). DNA demethylation pathways: Additional players and regulators. BioEssays, 39(1), 1–13. https://doi.org/10.1002/bies.201600178
Fan, H., Zhang, H., Pascuzzi, P. E., & Andrisani, O. (2016). Hepatitis B virus X protein induces EpCAM expression via active DNA demethylation directed by RelA in complex with EZH2 and TET2. Oncogene, 35(6), 715–726. https://doi.org/10.1038/onc.2015.122
Chakraborty, A., & Viswanathan, P. (2018). Methylation-demethylation dynamics: Implications of changes in acute kidney injury. Analytical Cellular Pathology, 2018, 8764384. https://doi.org/10.1155/2018/8764384. Amsterdam.
Weng, N. P., Araki, Y., & Subedi, K. (2012). The molecular basis of the memory T cell response: Differential gene expression and its epigenetic regulation. Nature Reviews Immunology, 12(4), 306–315. https://doi.org/10.1038/nri3173
Araki, Y., Fann, M., Wersto, R., & Weng, N. P. (2008). Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: Perforin and granzyme B). The Journal of Immunology, 180(12), 8102–8108. https://doi.org/10.4049/jimmunol.180.12.8102
Rodriguez, R. M., Suarez-Alvarez, B., Lavin, J. L., Mosen-Ansorena, D., Baragano Raneros, A., Marquez-Kisinousky, L., Aransay, A. M., & Lopez-Larrea, C. (2017). Epigenetic networks regulate the transcriptional program in memory and terminally differentiated CD8+ T cells. The Journal of Immunology, 198(2), 937–949. https://doi.org/10.4049/jimmunol.1601102
Maltby, V. E., Graves, M. C., Lea, R. A., Benton, M. C., Sanders, K. A., Tajouri, L., Scott, R. J., & Lechner-Scott, J. (2015). Genome-wide DNA methylation profiling of CD8+ T cells shows a distinct epigenetic signature to CD4+ T cells in multiple sclerosis patients. Clinical Epigenetics, 7, 118. https://doi.org/10.1186/s13148-015-0152-7
Vegran, F., Berger, H., Boidot, R., Mignot, G., Bruchard, M., Dosset, M., Chalmin, F., Rebe, C., Derangere, V., Ryffel, B., Kato, M., Prevost-Blondel, A., Ghiringhelli, F., & Apetoh, L. (2014). The transcription factor IRF1 dictates the IL-21-dependent anticancer functions of TH9 cells. Nature Immunology, 15(8), 758–766. https://doi.org/10.1038/ni.2925
Harrison, O. J., Linehan, J. L., Shih, H. Y., Bouladoux, N., Han, S. J., Smelkinson, M., Sen, S. K., Byrd, A. L., Enamorado, M., Yao, C., Tamoutounour, S., Van Laethem, F., Hurabielle, C., Collins, N., Paun, A., Salcedo, R., O’Shea, J. J., & Belkaid, Y. (2019). Commensal-specific T cell plasticity promotes rapid tissue adaptation to injury. Science, 363(6422). https://doi.org/10.1126/science.aat6280
Srenathan, U., Steel, K., & Taams, L. S. (2016). IL-17+ CD8+ T cells: Differentiation, phenotype and role in inflammatory disease. Immunology Letters, 178, 20–26. https://doi.org/10.1016/j.imlet.2016.05.001
Liu, Y., Yang, B., Ma, J., Wang, H., Huang, F., Zhang, J., Chen, H., & Wu, C. (2011). Interleukin-21 induces the differentiation of human Tc22 cells via phosphorylation of signal transducers and activators of transcription. Immunology, 132(4), 540–548. https://doi.org/10.1111/j.1365-2567.2010.03399.x
Motz, G. T., & Coukos, G. (2011). The parallel lives of angiogenesis and immunosuppression: Cancer and other tales. Nature Reviews Immunology, 11(10), 702–711. https://doi.org/10.1038/nri3064
Fox, C. J., Hammerman, P. S., & Thompson, C. B. (2005). Fuel feeds function: Energy metabolism and the T-cell response. Nature Reviews Immunology, 5(11), 844–852. https://doi.org/10.1038/nri1710
Jones, R. G., & Thompson, C. B. (2007). Revving the engine: Signal transduction fuels T cell activation. Immunity, 27(2), 173–178. https://doi.org/10.1016/j.immuni.2007.07.008
Buckley, A. F., Kuo, C. T., & Leiden, J. M. (2001). Transcription factor LKLF is sufficient to program T cell quiescence via a c-Myc-dependent pathway. Nature Immunology, 2(8), 698–704. https://doi.org/10.1038/90633
Brand, K. A., & Hermfisse, U. (1997). Aerobic glycolysis by proliferating cells: A protective strategy against reactive oxygen species. The FASEB Journal, 11(5), 388–395. https://doi.org/10.1096/fasebj.11.5.9141507
Frauwirth, K. A., Riley, J. L., Harris, M. H., Parry, R. V., Rathmell, J. C., Plas, D. R., Elstrom, R. L., June, C. H., & Thompson, C. B. (2002). The CD28 signaling pathway regulates glucose metabolism. Immunity, 16(6), 769–777. https://doi.org/10.1016/s1074-7613(02)00323-0
Christofk, H. R., Vander Heiden, M. G., Harris, M. H., Ramanathan, A., Gerszten, R. E., Wei, R., Fleming, M. D., Schreiber, S. L., & Cantley, L. C. (2008). The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature, 452(7184), 230–233. https://doi.org/10.1038/nature06734
DeBerardinis, R. J., Mancuso, A., Daikhin, E., Nissim, I., Yudkoff, M., Wehrli, S., & Thompson, C. B. (2007). Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A, 104(49), 19345–19350. https://doi.org/10.1073/pnas.0709747104
Facciabene, A., Peng, X., Hagemann, I. S., Balint, K., Barchetti, A., Wang, L. P., Gimotty, P. A., Gilks, C. B., Lal, P., Zhang, L., & Coukos, G. (2011). Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature, 475(7355), 226–230. https://doi.org/10.1038/nature10169
Shimizu, J., Yamazaki, S., & Sakaguchi, S. (1999). Induction of tumor immunity by removing CD25+CD4+ T cells: A common basis between tumor immunity and autoimmunity. The Journal of Immunology, 163(10), 5211–5218. https://www.ncbi.nlm.nih.gov/pubmed/10553041
Jiao, L., & Liu, X. (2015). Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science, 350(6258), aac4383. https://doi.org/10.1126/science.aac4383
O’Carroll, D., Erhardt, S., Pagani, M., Barton, S. C., Surani, M. A., & Jenuwein, T. (2001). The polycomb-group gene Ezh2 is required for early mouse development. Molecular and Cellular Biology, 21(13), 4330–4336. https://doi.org/10.1128/MCB.21.13.4330-4336.2001
Su, I. H., Basavaraj, A., Krutchinsky, A. N., Hobert, O., Ullrich, A., Chait, B. T., & Tarakhovsky, A. (2003). Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nature Immunology, 4(2), 124–131. https://doi.org/10.1038/ni876
Neo, W. H., Lim, J. F., Grumont, R., Gerondakis, S., & Su, I. H. (2014). c-Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells. Journal of Biological Chemistry, 289(46), 31693–31707. https://doi.org/10.1074/jbc.M114.574517
Cerboni, S., Jeremiah, N., Gentili, M., Gehrmann, U., Conrad, C., Stolzenberg, M. C., Picard, C., Neven, B., Fischer, A., Amigorena, S., Rieux-Laucat, F., & Manel, N. (2017). Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. Journal of Experimental Medicine, 214(6), 1769–1785. https://doi.org/10.1084/jem.20161674
Luksch, H., Stinson, W. A., Platt, D. J., Qian, W., Kalugotla, G., Miner, C. A., Bennion, B. G., Gerbaulet, A., Rosen-Wolff, A., & Miner, J. J. (2019). STING-associated lung disease in mice relies on T cells but not type I interferon. Journal of Allergy and Clinical Immunology, 144(1), 254-266 e258. https://doi.org/10.1016/j.jaci.2019.01.044
Yao, H., Wang, S., Zhou, X., Sun, J., Zhou, G., Zhou, D., Chen, G., Shi, X., Chen, J., Shi, B., Tai, Q., Mi, X., Sun, L., Yao, Y., & He, S. (2022). STING promotes proliferation and induces drug resistance in colorectal cancer by regulating the AMPK-mTOR pathway. Journal of Gastrointestinal Oncology, 13(5), 2458–2471. https://doi.org/10.21037/jgo-22-957
Domvri, K., Petanidis, S., Zarogoulidis, P., Anestakis, D., Tsavlis, D., Bai, C., Huang, H., Freitag, L., Hohenforst-Schmidt, W., Porpodis, K., & Katopodi, T. (2021). Treg-dependent immunosuppression triggers effector T cell dysfunction via the STING/ILC2 axis. Clinical Immunology, 222, 108620. https://doi.org/10.1016/j.clim.2020.108620
Wang, W., Kryczek, I., Dostal, L., Lin, H., Tan, L., Zhao, L., Lu, F., Wei, S., Maj, T., Peng, D., He, G., Vatan, L., Szeliga, W., Kuick, R., Kotarski, J., Tarkowski, R., Dou, Y., Rattan, R., Munkarah, A., & Zou, W. (2016). Effector T cells abrogate stroma-mediated chemoresistance in ovarian cancer. Cell, 165(5), 1092–1105. https://doi.org/10.1016/j.cell.2016.04.009
Xu, S., Tao, Z., Hai, B., Liang, H., Shi, Y., Wang, T., Song, W., Chen, Y., OuYang, J., Chen, J., Kong, F., Dong, Y., Jiang, S. W., Li, W., Wang, P., Yuan, Z., Wan, X., Wang, C., Li, W., & Chen, K. (2016). miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nature Communications, 7, 11406. https://doi.org/10.1038/ncomms11406
Zhang, L. N., Xin, T., Chen, M., & Gao, P. (2019). Chemoresistance in mesenchymal lung cancer cells is correlated to high regulatory T cell presence in the tumor microenvironment. IUBMB Life, 71(7), 986–991. https://doi.org/10.1002/iub.2043
Fife, B. T., & Bluestone, J. A. (2008). Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunological Reviews, 224, 166–182. https://doi.org/10.1111/j.1600-065X.2008.00662.x
Intlekofer, A. M., & Thompson, C. B. (2013). At the bench: Preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. Journal of Leukocyte Biology, 94(1), 25–39. https://doi.org/10.1189/jlb.1212621
Jain, N., Nguyen, H., Chambers, C., & Kang, J. (2010). Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proceedings of the National Academy of Sciences of the United States of America, 107(4), 1524–1528. https://doi.org/10.1073/pnas.0910341107
Takahashi, T., Tagami, T., Yamazaki, S., Uede, T., Shimizu, J., Sakaguchi, N., Mak, T. W., & Sakaguchi, S. (2000). Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. Journal of Experimental Medicine, 192(2), 303–310. https://doi.org/10.1084/jem.192.2.303
Walker, L. S. (2013). Treg and CTLA-4: Two intertwining pathways to immune tolerance. Journal of Autoimmunity, 45(100), 49–57. https://doi.org/10.1016/j.jaut.2013.06.006
Friedline, R. H., Brown, D. S., Nguyen, H., Kornfeld, H., Lee, J., Zhang, Y., Appleby, M., Der, S. D., Kang, J., & Chambers, C. A. (2009). CD4+ regulatory T cells require CTLA-4 for the maintenance of systemic tolerance. Journal of Experimental Medicine, 206(2), 421–434. https://doi.org/10.1084/jem.20081811
Grosso, J. F., & Jure-Kunkel, M. N. (2013). CTLA-4 blockade in tumor models: An overview of preclinical and translational research. Cancer Immunity, 13, 5. https://www.ncbi.nlm.nih.gov/pubmed/23390376
Ishida, Y., Agata, Y., Shibahara, K., & Honjo, T. (1992). Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO Journal, 11(11), 3887–3895. https://doi.org/10.1002/j.1460-2075.1992.tb05481.x
Keir, M. E., Butte, M. J., Freeman, G. J., & Sharpe, A. H. (2008). PD-1 and its ligands in tolerance and immunity. Annual Review of Immunology, 26, 677–704. https://doi.org/10.1146/annurev.immunol.26.021607.090331
He, Y. F., Zhang, G. M., Wang, X. H., Zhang, H., Yuan, Y., Li, D., & Feng, Z. H. (2004). Blocking programmed death-1 ligand-PD-1 interactions by local gene therapy results in enhancement of antitumor effect of secondary lymphoid tissue chemokine. The Journal of Immunology, 173(8), 4919–4928. https://doi.org/10.4049/jimmunol.173.8.4919
Hirano, F., Kaneko, K., Tamura, H., Dong, H., Wang, S., Ichikawa, M., Rietz, C., Flies, D. B., Lau, J. S., Zhu, G., Tamada, K., & Chen, L. (2005). Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Research, 65(3), 1089–1096. https://www.ncbi.nlm.nih.gov/pubmed/15705911
Iwai, Y., Ishida, M., Tanaka, Y., Okazaki, T., Honjo, T., & Minato, N. (2002). Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A, 99(19), 12293–12297. https://doi.org/10.1073/pnas.192461099
Iwai, Y., Terawaki, S., & Honjo, T. (2005). PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. International Immunology, 17(2), 133–144. https://doi.org/10.1093/intimm/dxh194
Foster, R., Buckanovich, R. J., & Rueda, B. R. (2013). Ovarian cancer stem cells: Working towards the root of stemness. Cancer Letters, 338(1), 147–157. https://doi.org/10.1016/j.canlet.2012.10.023
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J., & Schreiber, R. D. (2002). Cancer immunoediting: From immunosurveillance to tumor escape. Nature Immunology, 3(11), 991–998. https://doi.org/10.1038/ni1102-991
Teng, M. W., Galon, J., Fridman, W. H., & Smyth, M. J. (2015). From mice to humans: Developments in cancer immunoediting. The Journal of Clinical Investigation, 125(9), 3338–3346. https://doi.org/10.1172/JCI80004
McGranahan, N., Rosenthal, R., Hiley, C. T., Rowan, A. J., Watkins, T. B. K., Wilson, G. A., Birkbak, N. J., Veeriah, S., Van Loo, P., Herrero, J., Swanton, C., & Consortium, T. R. (2017). Allele-specific HLA loss and immune escape in lung cancer evolution. Cell, 171(6), 1259–1271 e1211. https://doi.org/10.1016/j.cell.2017.10.001
Sade-Feldman, M., Jiao, Y. J., Chen, J. H., Rooney, M. S., Barzily-Rokni, M., Eliane, J. P., Bjorgaard, S. L., Hammond, M. R., Vitzthum, H., Blackmon, S. M., Frederick, D. T., Hazar-Rethinam, M., Nadres, B. A., Van Seventer, E. E., Shukla, S. A., Yizhak, K., Ray, J. P., Rosebrock, D., Livitz, D., & Hacohen, N. (2017). Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nature Communications, 8(1), 1136. https://doi.org/10.1038/s41467-017-01062-w
Saleh, R., & Elkord, E. (2020). Acquired resistance to cancer immunotherapy: Role of tumor-mediated immunosuppression. Seminars in Cancer Biology, 65, 13–27. https://doi.org/10.1016/j.semcancer.2019.07.017
Taylor, A., Verhagen, J., Blaser, K., Akdis, M., & Akdis, C. A. (2006). Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: The role of T regulatory cells. Immunology, 117(4), 433–442. https://doi.org/10.1111/j.1365-2567.2006.02321.x
Voron, T., Marcheteau, E., Pernot, S., Colussi, O., Tartour, E., Taieb, J., & Terme, M. (2014). Control of the immune response by pro-angiogenic factors. Frontiers in Oncology, 4, 70. https://doi.org/10.3389/fonc.2014.00070
Schatton, T., Schutte, U., Frank, N. Y., Zhan, Q., Hoerning, A., Robles, S. C., Zhou, J., Hodi, F. S., Spagnoli, G. C., Murphy, G. F., & Frank, M. H. (2010). Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Research, 70(2), 697–708. https://doi.org/10.1158/0008-5472.CAN-09-1592
Luo, N., Nixon, M. J., Gonzalez-Ericsson, P. I., Sanchez, V., Opalenik, S. R., Li, H., Zahnow, C. A., Nickels, M. L., Liu, F., Tantawy, M. N., Sanders, M. E., Manning, H. C., & Balko, J. M. (2018). DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nature Communications, 9(1), 248. https://doi.org/10.1038/s41467-017-02630-w
Wylie, B., Chee, J., Forbes, C. A., Booth, M., Stone, S. R., Buzzai, A., Abad, A., Foley, B., Cruickshank, M. N., & Waithman, J. (2019). Acquired resistance during adoptive cell therapy by transcriptional silencing of immunogenic antigens. Oncoimmunology, 8(8), 1609874. https://doi.org/10.1080/2162402X.2019.1609874
Burr, M. L., Sparbier, C. E., Chan, K. L., Chan, Y. C., Kersbergen, A., Lam, E. Y. N., Azidis-Yates, E., Vassiliadis, D., Bell, C. C., Gilan, O., Jackson, S., Tan, L., Wong, S. Q., Hollizeck, S., Michalak, E. M., Siddle, H. V., McCabe, M. T., Prinjha, R. K., Guerra, G. R., & Dawson, M. A. (2019). An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell, 36(4), 385-401 e388. https://doi.org/10.1016/j.ccell.2019.08.008
Siebenkas, C., Chiappinelli, K. B., Guzzetta, A. A., Sharma, A., Jeschke, J., Vatapalli, R., Baylin, S. B., & Ahuja, N. (2017). Inhibiting DNA methylation activates cancer testis antigens and expression of the antigen processing and presentation machinery in colon and ovarian cancer cells. PLoS ONE, 12(6), e0179501. https://doi.org/10.1371/journal.pone.0179501
Gameiro, S. R., Malamas, A. S., Tsang, K. Y., Ferrone, S., & Hodge, J. W. (2016). Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget, 7(7), 7390–7402. https://doi.org/10.18632/oncotarget.7180
Lo Re, O., Mazza, T., Giallongo, S., Sanna, P., Rappa, F., Vinh Luong, T., Li Volti, G., Drovakova, A., Roskams, T., Van Haele, M., Tsochatzis, E., & Vinciguerra, M. (2020). Loss of histone macroH2A1 in hepatocellular carcinoma cells promotes paracrine-mediated chemoresistance and CD4(+)CD25(+)FoxP3(+) regulatory T cells activation. Theranostics, 10(2), 910–924. https://doi.org/10.7150/thno.35045
Mazzone, R., Zwergel, C., Mai, A., & Valente, S. (2017). Epi-drugs in combination with immunotherapy: A new avenue to improve anticancer efficacy. Clinical Epigenetics, 9, 59. https://doi.org/10.1186/s13148-017-0358-y
Grinberg-Bleyer, Y., Oh, H., Desrichard, A., Bhatt, D. M., Caron, R., Chan, T. A., Schmid, R. M., Klein, U., Hayden, M. S., & Ghosh, S. (2017). NF-kappaB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell, 170(6), 1096-1108 e1013. https://doi.org/10.1016/j.cell.2017.08.004
Xiao, G., Jin, L. L., Liu, C. Q., Wang, Y. C., Meng, Y. M., Zhou, Z. G., Chen, J., Yu, X. J., Zhang, Y. J., Xu, J., & Zheng, L. (2019). EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. Journal for Immunotherapy of Cancer, 7(1), 300. https://doi.org/10.1186/s40425-019-0784-9
Wang, D., Quiros, J., Mahuron, K., Pai, C. C., Ranzani, V., Young, A., Silveria, S., Harwin, T., Abnousian, A., Pagani, M., Rosenblum, M. D., Van Gool, F., Fong, L., Bluestone, J. A., & DuPage, M. (2018). Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Reports, 23(11), 3262–3274. https://doi.org/10.1016/j.celrep.2018.05.050
Goswami, S., Apostolou, I., Zhang, J., Skepner, J., Anandhan, S., Zhang, X., Xiong, L., Trojer, P., Aparicio, A., Subudhi, S. K., Allison, J. P., Zhao, H., & Sharma, P. (2018). Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. The Journal of Clinical Investigation, 128(9), 3813–3818. https://doi.org/10.1172/JCI99760
DuPage, M., Chopra, G., Quiros, J., Rosenthal, W. L., Morar, M. M., Holohan, D., Zhang, R., Turka, L., Marson, A., & Bluestone, J. A. (2015). The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity, 42(2), 227–238. https://doi.org/10.1016/j.immuni.2015.01.007
Woo, E. Y., Chu, C. S., Goletz, T. J., Schlienger, K., Yeh, H., Coukos, G., Rubin, S. C., Kaiser, L. R., & June, C. H. (2001). Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Research, 61(12), 4766–4772. https://www.ncbi.nlm.nih.gov/pubmed/11406550
Li, X., Xing, J., Wang, H., & Yu, E. (2019). The SLC34A2-ROS-HIF-1-induced up-regulation of EZH2 expression promotes proliferation and chemo-resistance to apoptosis in colorectal cancer. Bioscience Reports, 39(5). 10.1042/BSR20180268
Yang, Q., Zhao, S., Shi, Z., Cao, L., Liu, J., Pan, T., Zhou, D., & Zhang, J. (2021). Chemotherapy-elicited exosomal miR-378a-3p and miR-378d promote breast cancer stemness and chemoresistance via the activation of EZH2/STAT3 signaling. Journal of Experimental & Clinical Cancer Research, 40(1), 120. https://doi.org/10.1186/s13046-021-01901-1
Gardner, E. E., Lok, B. H., Schneeberger, V. E., Desmeules, P., Miles, L. A., Arnold, P. K., Ni, A., Khodos, I., de Stanchina, E., Nguyen, T., Sage, J., Campbell, J. E., Ribich, S., Rekhtman, N., Dowlati, A., Massion, P. P., Rudin, C. M., & Poirier, J. T. (2017). Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 axis. Cancer Cell, 31(2), 286–299. https://doi.org/10.1016/j.ccell.2017.01.006
Zhan, J., Wang, P., Li, S., Song, J., He, H., Wang, Y., Liu, Z., Wang, F., Bai, H., Fang, W., Du, Q., Ye, M., Chang, Z., Wang, J., & Zhang, H. (2019). HOXB13 networking with ABCG1/EZH2/Slug mediates metastasis and confers resistance to cisplatin in lung adenocarcinoma patients. Theranostics, 9(7), 2084–2099. https://doi.org/10.7150/thno.29463
Chen, X., Xie, R., Gu, P., Huang, M., Han, J., Dong, W., Xie, W., Wang, B., He, W., Zhong, G., Chen, Z., Huang, J., & Lin, T. (2019). Long noncoding RNA LBCS inhibits self-renewal and chemoresistance of bladder cancer stem cells through epigenetic silencing of SOX2. Clinical Cancer Research, 25(4), 1389–1403. https://doi.org/10.1158/1078-0432.CCR-18-1656
Wang, C., Li, X., Zhang, J., Ge, Z., Chen, H., & Hu, J. (2018). EZH2 contributes to 5-FU resistance in gastric cancer by epigenetically suppressing FBXO32 expression. Oncotargets and Therapy, 11, 7853–7864. https://doi.org/10.2147/OTT.S180131
Ye, K., & Wang, Y. (2022). Long non-coding RNA ZNF674-AS1 antagonizes oxaliplatin resistance of gastric cancer via regulating EZH2-mediated methylation of CHST7. Aging (Albany NY), 14(13), 5523–5536. https://doi.org/10.18632/aging.204165
Zhou, J., Che, J., Xu, L., Yang, W., Li, Y., Zhou, W., & Zou, S. (2022). Enhancer of zeste homolog 2 promotes hepatocellular cancer progression and chemoresistance by enhancing protein kinase B activation through microRNA-381-mediated SET domain bifurcated 1. Bioengineered, 13(3), 5737–5755. https://doi.org/10.1080/21655979.2021.2023792
Yang, L. H., Du, P., Liu, W., An, L. K., Li, J., Zhu, W. Y., Yuan, S., Wang, L., & Zang, L. (2021). LncRNA ANRIL promotes multiple myeloma progression and bortezomib resistance by EZH2-mediated epigenetically silencing of PTEN. Neoplasma, 68(4), 788–797. https://doi.org/10.4149/neo_2021_210205N184
Funding
This work was supported by Basic and Applied Research in Biophysics and Material Science (RSI 4002) by the Department of Atomic Energy (DAE), S. Ramachandran National Bioscience Award for Career Development 2019 (BT/HRD-NBA-NWB/38/2019-20), Department of Biotechnology, and SwarnaJayanti Fellowship (DST/SJF/LSA-02/2017-18), Department of Science and Technology, Govt. of India to C. D.
Author information
Ethics declarations
Ethical approval
The authors have adhered to all pertinent ethical standards while drafting this manuscript. They have taken all required measures to guarantee the precision and comprehensiveness of the provided information.
Informed consent
N/A.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Singh, V., Nandi, S., Ghosh, A. et al. Epigenetic reprogramming of T cells: unlocking new avenues for cancer immunotherapy. Cancer Metastasis Rev 43, 175–195 (2024). https://doi.org/10.1007/s10555-024-10167-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-024-10167-w