
Abstract
Recurrent, clonal somatic mutations in histone H3 are molecular hallmarks that distinguish the genetic mechanisms underlying pediatric and adult high-grade glioma (HGG), define biological subgroups of diffuse glioma, and highlight connections between cancer, development, and epigenetics. These oncogenic mutations in histones, now termed “oncohistones”, were discovered through genome-wide sequencing of pediatric diffuse high-grade glioma. Up to 80% of diffuse midline glioma (DMG), including diffuse intrinsic pontine glioma (DIPG) and diffuse glioma arising in other midline structures including thalamus or spinal cord, contain histone H3 lysine 27 to methionine (K27M) mutations or, rarely, other alterations that result in a depletion of H3K27me3 similar to that induced by H3 K27M. This subgroup of glioma is now defined as diffuse midline glioma, H3K27-altered. In contrast, histone H3 Gly34Arg/Val (G34R/V) mutations are found in approximately 30% of diffuse glioma arising in the cerebral hemispheres of older adolescents and young adults, now classified as diffuse hemispheric glioma, H3G34-mutant. Here, we review how oncohistones modulate the epigenome and discuss the mutational landscape and invasive properties of histone mutant HGGs of childhood. The distinct mechanisms through which oncohistones and other mutations rewrite the epigenetic landscape provide novel insights into development and tumorigenesis and may present unique vulnerabilities for pHGGs. Lessons learned from these rare incurable brain tumors of childhood may have broader implications for cancer, as additional high- and low-frequency oncohistone mutations have been identified in other tumor types.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Diffusely infiltrative pediatric high-grade gliomas (pHGGs) are among childhood's most devasting central nervous system (CNS) malignancies. Despite accounting for only 15–20% of all childhood brain tumors, pHGGs are the leading cause of cancer-related morbidity and mortality in children [1, 2]. To improve the therapeutic outlook for these devastating tumors, researchers mapped the molecular and genomic landscape of pHGGs. Independent sequencing efforts of primary patient samples converged on a remarkable finding: high frequency recurrent missense mutations in histone H3 at amino acid 27 (lysine to methionine; H3 K27M) in diffuse intrinsic pontine glioma (DIPG) and other diffuse midline gliomas (DMG) and at amino acid 34 (glycine to arginine/valine; H3 G34R/V) in gliomas arising in the cerebral hemispheres [3,4,5]. Alterations in histone H3 K27 or G34, with mutually exclusive mutational patterns, were identified in more than 50% of pHGGs and are now considered a defining molecular feature of subgroups of childhood glioma. In contrast to HGGs in children, less than 1% of adult HGGsharbor these mutations [6, 7].
These “oncohistones” were the first histone mutations identified in human cancer. A wide range of cancer mutations had been identified previously in enzymes that added, removed, or selectively bound to post-translational modifications on histones; however, it was unclear if these mutations only influenced modifications or binding to other cellular proteins unrelated to chromatin. Mutation of histone H3 itself, a core component of the nucleosome, provided incontrovertible genetic evidence for a central role of disrupted epigenetic regulation in cancer and demonstrated distinct selective pressures and etiology driving childhood and adult high-grade glioma.
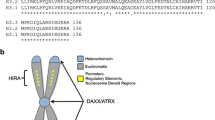
There are three isoforms of histone H3 that are encoded by a multigene family of 15 genes. H3.1 and H3.2 are the “canonical histones” with replication-dependent peak expression during S-phase to generate the necessary increase in nucleosomes to package newly replicated DNA [8]. The “non-canonical” or replication-independent histone H3.3 is expressed and incorporated into nucleosomes independent of the cell cycle and is involved in regulation and maintenance of chromatin environments [9]. Only five amino acid residues differentiate histones H3.1 and H3.2 from histone H3.3, and it is believed that three of those residues (87, 89 and 90) are responsible for conferring chaperone specificity to each of these closely related proteins (Fig. 1a) [9, 10]. H3.1 and H3.2 are deposited during S phase via the CAF1 complex (Fig. 1b) [10], while H3.3 is incorporated selectively into “open” chromatin, such as transcriptionally active regions, via the HIRA complex and into heterochromatic regions by DAXX-ATRX (Fig. 1c) [11]. Additionally, H3.3 and ATRX contribute to telomere maintenance in embryonic stem and differentiated cells [11,12,13]. These differences in nucleosome assembly at specific points during development result in distinct deposition patterns with broad expression of H3.1/2 across the genome and enrichment of H3.3 at specific loci (Fig. 1d) [8].

Canonical and non-canonical histone H3 are differentially regulated during development. a) Schematic illustrating the five amino acids that differ between histones H3.1, H3.2 and H3.3 (white font in red squares). Frequently mutated sites are denoted with asterisks. Histones b) H3.1/2 and c) H3.3 are deposited into nucleosomes at different genomic locations and at different points in the cell cycle through distinct chaperones. d) Schematic distribution of histones H3.1/2 (light green) and H3.3 (dark green) across chromosomes demonstrate enrichment at specific genomic sites and DNA processes (figure adapted from [14]). H3.1 is broadly deposited throughout the chromosome while H3.3 deposition is enriched at telomeres, centromeres, regulatory elements, actively transcribed regions, and in regions that have undergone DNA repair
H3 K27M mutation is found in up to 80% of diffuse midline glioma (DMG), arising in midline brain structures of children, including the brainstem, pons (diffuse intrinsic pontine glioma; DIPG), thalamus, and more rarely, the cerebellum and spinal cord [3,4,5, 15]. Despite the high level of redundancy in the genes encoding histone H3, approximately 75% of K27M mutations occur in H3F3A, one of two genes encoding H3.3. The remaining one-quarter of mutations occur in one of ten genes encoding the replication-dependent H3.1 variant, most commonly HIST1H3B, and rarely in other genes encoding H3.1 or H3.2 [7]. Despite being broadly expressed throughout the brain [16], tumors driven by H3.1/2 or H3.3 arise at slightly different ages and locations. H3.1 K27M tumors almost exclusively occur in the pons in the youngest DMG patients, with a median age of approximately 5 years old and no difference in sex stratification, while H3.3 K27M tumors span a broader range of midline structures and ages with a median age of onset of approximately 8 years old [3, 5, 15, 17,18,19]. Tumors driven by K27M mutations are almost universally fatal, with most patients succumbing to their disease within 3 years of diagnosis (Fig. 2) [7, 20].

Overview of pediatric high-grade gliomas. a) Graphs depicting the age of onset in years across pediatric high-grade glioma subgroups with the median age for each sex denoted above the graph and the percentage of patients of each sex denoted below the graph. The dotted horizontal line represents the threshold for pediatric classification at 21 years old. The asterisks indicate significant sex differences. b) Cartoon brains illustrate common tumor locations (marked in shades of red) within each subgroup. The darker shades indicate regions with higher incidence for the associated subgroup. c) Graphs depicting the most common non-synonymous genetic mutations associated with each tumor subgroup, excluding amplifications and losses. d) Kaplan–Meier curves depicting overall survival rates across subgroups with the median survival indicated in months in the top right corner of each graph. Graphs and survival curves generated with published data [4, 6, 7, 18, 21,22,23,24]
In contrast, H3 G34R/V mutations have been identified exclusively in H3.3 encoded by H3F3A. H3 G34R/V mutations are found in diffusely infiltrative tumors, almost always arising in the cerebral hemispheres, predominantly in the parietal or temporal lobes of adolescents and young adults, with a median age of 15–19 years and a male predominance (M:F ratio of 1.4:1). Despite a paucity of literature characterizing this tumor entity, reports estimate that more than 30% of hemispheric pHGGs bear H3.3 G34R/V mutations [3,4,5, 7, 21, 25]. While patients with H3 G34 mutations exhibit prolonged survival when compared to patients with H3 K27M mutations, overall survival remains limited to 5 years after diagnosis [7]. In contrast, IDH-mutant astrocytoma, which overlaps in age with G34-mutant glioma, presents mainly in forebrain and exhibits a median overall survival of approximately 38 months after diagnosis (Fig. 2) [4, 7, 26].
H3- and IDH-wildtype diffuse pHGGs and infant-type hemispheric gliomas (IHG) lack the characteristic oncohistone mutations found in other diffuse midline and hemispheric gliomas. IHGs occur in the youngest patients, with a median age of 2.8 months at time of presentation [22, 27]. Up to 80% of these tumors harbor gene fusions in which one of the fusion partners is a receptor tyrosine kinase (RTK) such as NTRK, ALK, ROS1, or MET [17, 22, 27]. H3- and IDH-WT diffuse pHGGs have a broader distribution across ages, occurring in children, adolescents, and young adults [23]. These tumors are mostly supratentorial but have also been reported in midline structures [7, 17], and they typically harbor amplifications or mutations in PDGFRA, TP53, NF1, EGFR or MYCN (Fig. 2) [28].
The intriguing spatiotemporal incidence of oncohistone mutations suggests that specific mutations confer unique selective advantages in distinct developmental settings. Thus, pHGG affords a physiologically relevant opportunity to study the intricate relationship between epigenetic dysregulation, disrupted development, and tumorigenesis.
2 Epigenetic consequences of oncohistone mutations
2.1 Dominant negative effect of H3 K27M on PRC2
Oncohistone mutations like H3 K27M likely prime cells for oncogenic transformation through genome-wide epigenetic changes. Although H3 K27M comprises only 5–17% of the total H3 expressed, it exerts a potent dominant negative effect through multiple mechanisms to inhibit polycomb repressive complex 2 (PRC2), which deposits methyl marks on histone H3 at lysine 27. This effect of the H3 K27M mutation in the minority of cellular H3 results in a dramatic global loss of the repressive H3K27 tri-methyl (H3K27me3) mark on the large cellular pool of wild-type H3(Fig. 3a) [29,30,31,32]. Studies performed in immortalized HEK 293 T cells were among the first to show that the K27M mutation in histone H3 specifically represents a gain-of-function mutation, potentially reducing di- and tri-methylation at H3K27 by targeting the active site in the SET domain-containing methyltransferase EZH2, the catalytic component of PRC2 [32]. H3 K27M also exhibits a higher binding affinity to PRC2 than wildtype H3 K27 and thus may preferentially bind and sequester PRC2, limiting the availability of functional PRC2 [32,33,34]. ChIP-seq analyses from K27M mutant glioma cells showed that PRC2 components did not colocalize with H3.3 K27M occupancy, arguing against a long-lived sequestration as the primary mechanism of PRC2 inhibition [35]. Interestingly, upon release from H3 K27M, PRC2 loses histone methyltransferase activity, potentially through an H3 K27M-driven lasting conformational change [36,37,38]. H3 K27M expression also reduces EZH2 automethylation, impairing PRC2 activity [39,40,41], and resulting in loss of H3K27me3 spreading along the genome [42]. Together, these studies indicate that H3 K27M oncohistones impair the intrinsic activity of PRC2 to prevent H3K27 methylation.

Oncohistones dysregulate histone methylation patterns. a) PRC2 deposits methyl groups (red circles) at H3K27 in wild-type cells. H3 K27M causes global loss of H3K27me3 while H3 G34R/V increases H3K27me3 at active enhancers. b) SETD2 and SETD5 deposit the third methyl group at H3K36 (dark red circles), which can be removed by KDM4 (KDM4A, KDM4B, and KDM4D). H3K36me3 is unaffected in H3 K27-altered tumors while H3 G34R/V causes a loss of H3K36me3 in cis
Of note, PRC2 loss of function, through inactivating mutation in essential components such as EZH2, EED, or SUZ12, is found in other cancers like early T-progenitor ALL, and leads to a more complete loss of H3K27me3 than H3 K27M-mediated PRC2 dysfunction [43]. Genomic aberrations leading to PRC2 loss of function are not observed in DMG, however, which suggests H3 K27M-mediated epigenetic dysregulation generates a uniquely permissive and advantageous epigenetic environment for gliomagenesis involving significant but not complete loss of PRC2.
2.2 Retention of H3K27me3
While H3 K27M causes a global loss of H3K27me3, some regions, especially in strong PRC2 targets, retain or gain this repressive mark (Fig. 4a). These regions include genes that are involved in developmental regulation, including patterning, Hox genes (developmental identity), Cdkn2a (tumor suppressor), and neurogenesis with the specific targets of repression varying depending on cell states and spatial origin [35, 40, 42, 44,45,46]. However, there were also examples in which enhancers showed H3 K27M occupancy accompanied by increased PRC2 binding and H3K27me3 deposition, and decreased expression of neurodevelopmental genes [34, 44]. Focal H3K27me3 retention may thus serve multiple purposes. For example, H3 K27M may reinforce cellular states or identities by repressing loci related to cellular origins while also inhibiting tumor suppressor and alternative differentiation mechanisms. This indicates that H3K27me3-retained genes may represent genes imperative for tumorigenic function and explain why more complete PRC2 loss of function mutations are not found in DMG. In fact, EZH2 inhibition or loss of function can selectively restore expression of genes aberrantly silenced by H3 K27M and decrease survival of H3 K27M DMG cells [35, 44, 46].

Oncohistones produce widespread epigenetic consequences. H3 K27-altered tumors exhibit a) altered deposition of specific epigenetic marks, b) activation of bivalent genes and c) reactivation of endogenous retroviral elements. a) H3 K27-altered tumors demonstrate global loss of H3K27me3 (left) but focally retain the mark at specific loci. Conversely, H3 K27-altered tumors exhibit spreading of H3K36me2 (right) across the genome. b) Bivalent promoters are characterized by the combined presence of the activating mark H3K4me3 (blue diamond) and the repressive mark H3K27me3 (red circle) and are poised for induced gene expression. The global loss of H3K27me3 in H3 K27-altered tumors releases bivalency and activates gene expression. H3 K27M may also recruit MLL1 to deposit H3K4me3 at distal regions. c) Repeat elements, such as endogenous retroviral elements (ERVs) normally retain H3K27me3 and are not expressed. H3 K27-altered tumors promote CBP/P300 recruitment at ERVs, resulting in H3K27ac (dark blue circle) deposition and priming ERVs for expression
Although the primary epigenetic consequence of H3 K27M is widespread loss of H3K27me3, H3 K27M also affects H3K36 methylation. H3K36 trimethylation (H3K36me3) is an epigenetic mark associated with actively expressed genes and transcriptional elongation [47]. H3K36 di- and trimethylation (H3K36me2/me3) appears to antagonize H3K27me3 and may function as a boundary to prevent the spread of H3K27me3 across the genome [48, 49]. H3 K27M results in spreading of H3K36me2/me3 to occupy regions previously associated with H3K27me3 (Fig. 4a) [38, 42, 50], but does not induce a significant change in the global levels of H3K36me2/me3 as measured by quantitative mass spectrometry [32].
2.3 Bivalency
The regulation of H3K27me3 is particularly important for ‘bivalent’ genes, which play key roles in development. Bivalent genes harbor both the repressive H3K27me3 mark and the active H3K4me3 mark at their promoter. Bivalent genes are said to be “poised” as they are transcriptionally silent but ready to be rapidly switched on. Such genes are “released” during development and cell-state transitions when H3K27me3 is lost. The global loss of H3K27me3 in K27M-mutant tumors causes dysregulation of genes with bivalent promoters [45, 51, 52]. Transcriptionally upregulated genes in a mouse model of H3 K27M DMG compared to WT DMG were significantly enriched for genes with bivalent promoters [45]. Additionally, in DMG patient-derived xenografts (PDXs), H3 K27M promoted tumor growth by maintaining cells in a progenitor-like epigenetic state through the reduction of H3K27me3 at bivalent promoters, increasing the expression of factors associated with cycling and stem-like cells [52]. Together, these studies show H3 K27M can release bivalent promoters through the loss of H3K27me3. H3 K27M can also directly recruit MLL1, the histone methyltransferase responsible for H3K4 methylation, to influence aberrant distribution of H3K4me3 deposition. This occurs primarily at distal regions, altering H3K4me3 distribution and leading to unbalanced bivalent chromatin [53]. These studies provide a mechanism by which H3 K27M perturbs gene expression by disrupting bivalent chromatin states (Fig. 4b).
2.4 Altered active chromatin landscape
The global loss of H3K27me3 coincides with a reciprocal global increase in H3K27 acetylation (H3K27ac) [32, 45, 52, 54]. While this increase is modest, it may have relevant biological impact. Expression of H3 K27M in patient-derived HGG cell lines causes aberrant deposition of H3K27ac, which is enriched at endogenous retroviral elements (ERVs), correlates with increased expression, and may play a role in cancer through viral mimicry (Fig. 4c) [54, 55]. H3K27ac also appears to be enriched in H3 K27M-containing nucleosomes which colocalize with bromodomain-containing proteins BRD2/4, key regulators of RNA Polymerase II, at actively transcribed genes [35]. This suggests that increased H3K27ac may promote aberrant transcriptional activation through recruitment of transcriptional activators. BRD inhibition with JQ1 was sufficient to induce growth arrest and increase expression of neuronal signatures in patient-derived H3 K27M DMG cell lines [35], illustrating a functional role for aberrant transcriptional activation in proliferation and inhibition of differentiation.
Super-enhancers (SEs), large groups of enhancers heavily bound by multiple regulatory TFs, have been strongly linked to oncogenic transformation and maintenance of tumor cell states [56]. H3 K27M HGGs exhibit distinct SE signatures when compared to H3 WT HGGs and other pediatric brain tumors. Genes regulated by these unique SE landscapes include distinct H3 K27M signature genes such as LIN28B, genes related to regional identity including PAX3 and IRX2, the stemness factor SOX2, and oligodendrocyte precursor cell (OPC)/ oligodendrocyte (OL)-lineage regulators such as PDGFRA, OLIG2, and MYRF. Signal transduction pathways, like Notch, EGFR, and p38 MAPK are also enriched in SE-associated genes and contribute to oncogenic function in H3 K27M DMG by increasing cell viability, migration, and invasion [57]. Therefore, genes associated with SEs may represent a core set of transcriptional programs important for DMG identity and malignant state.
In patient tumors, oncohistone H3.1 K27M and H3.3 K27M deposition mirrors the pattern of their wildtype counterparts that is shown in Fig. 1 [40, 58]. Because H3.1 K27M is broadly deposited across the genome, it is clear that K27M-induced transcriptional changes are not simply regulated by the location of oncohistone deposition. Expression of H3.3 K27M or H3.1 K27M in human induced oligodendrocyte progenitor cells establishes distinct enhancer landscapes resembling DMG with the respective mutations, while expression in induced neural stem cells resulted in enhancer landscapes that were less similar to DMG [58]. This illustrates that the overall landscape and similarity to patient tumors is influenced by both the H3 variant of K27M and by the cell state in which they are expressed, reflecting the critical interplay between oncohistones, transformation, and developmental context.
2.5 Epigenetic dysregulation in H3.3 G34R/V
Despite the genetic evidence from patient tumors that only one H3.3 G34R/V mutant allele is needed in the genesis of gliomas bearing this mutation, mechanistic data demonstrating a dominant functional mechanism of action of H3.3 G34R/V is lacking. H3.3 G34R/V mutations disrupt di- and trimethylation of the adjacent H3K36 (H3K36me2/3) due to steric hindrance of SETD2, a lysine 36 methyltransferase [34, 59, 60]. This is reminiscent of a subset of hemispheric gliomas in which SETD2 loss of function mutations confer a global loss of H3K36me3 [61, 62]. However, H3.3 G34R/V mutations only induce a loss of H3K36me2/3 in cis on those histones harboring the H3 G34R/V mutation, and an accompanying increase in PRC2 activity that may play a role in enhancer dysfunction (Fig. 3b) [32, 63].
Significant efforts have been made toward characterizing the impact of the H3.3 G34R/V mutation on the epigenome. Of particular interest is how this mutation may cooperate with other factors and lineage of origin spatiotemporal gene regulatory programs to promote gliomagenesis [21, 32, 60, 64,65,66,67,68]. In a single patient-derived cell line with an H3.3 G34V mutation, MYCN was found to be highly upregulated and was the most differentially bound gene by H3K36me3. These findings suggest that the proto-oncogene MYCN could potentially drive gliomagenesis in H3 G34-mutant tumors [69]. However, further validation using additional models is critically needed. Apart from patient-derived models, studies using mouse embryonic stem cells engineered to express the H3.3 G34R mutation exhibited increased levels of H3K9me3 and H3K36me3 at loci enriched for binding by the H3K9/H3K36 demethylase, KDM4 [68]. This may support a mechanism similar to that of SETD2 mutations by which the H3.3 G34R mutation inhibits KDM4 activity by impairing access to K36. Lastly, genomic instability resulting from impaired DNA repair mechanisms has also been implicated as a potential contributor to H3.3 G34R/V tumorigenesis [59, 65].
2.6 Epigenetic alterations in H3 WT HGGs
While IHGs appear to lack mutations driving epigenetic dysregulation characteristic of H3 K27-altered and H3 G34-mutant pHGGs, altered epigenetic landscapes have been observed in H3- and IDH-WT diffuse pHGGs. These include amplification of the lysine demethylase KDM6A, deletions or truncations in the transcriptional co-repressors BCOR and BCORL1, truncation of the histone acetyltransferase CREBBP, and truncation of the transcriptional regulator LZTR1 [70]. Truncating alterations in the H3K36 trimethylase, SETD2, are of particular interest as they rarely co-occur with oncohistone mutations and have been observed almost exclusively in hemispheric tumors in older pediatric patients [62, 71]. SETD2 mutations result in decreased H3K36me3, similarly to H3.3 G34R/V mutations [32, 62, 71], but demonstrate epigenetic signatures more closely resembling those found in IDH mutant tumors [62]. Somatic mutations in other histone writers or erasers and chromatin remodeling genes are common in pHGG and occur in H3 WT and histone mutant HGGs. Although specific individual genes were not mutated at high frequency, collectively 22% of DMGs and 48% of cortical pHGGs contained mutations in epigenetic regulators other than histone mutations [17]. In a large cohort of adult glioblastoma, concomitant mutation of IHD1 and ATRX were recurrent among this subgroup of tumors. More broadly, mutually exclusive mutations in genes involved in chromatin modification including MLL family members and other histone methyltransferases, histone deacetylases, histone demethylases, and SWI/SNF complex members, were identified in 46% of patients suggesting contributions of chromatin deregulation in glioma pathobiology across different age groups [61].
Thus, genetic and epigenetic dysregulation, whether through the expression of oncohistones or other mutations, may prime cells during development for oncogenic transformation. IHG may arise from more primitive progenitor cells in which mutations are not required to establish a permissive epigenetic state.
3 Oncohistones define new glioma classifications
Concerted efforts focusing on mutational status, tumor location, histological findings, age of onset, and DNA methylation signatures have refined brain tumor classifications and prompted the World Health Organization to establish new glioma subtype classifications. Histone mutations represent defining molecular hallmarks and demonstrate striking anatomic and age-dependent selectivity resulting in the new classifications of diffuse midline glioma, H3 K27-altered and diffuse hemispheric glioma, H3 G34-mutant [72].
3.1 DNA methylation signatures of brain tumor subgroups
DNA methylation profiling may reflect cell state, anatomic location, and tumor-specific signatures, and has become a powerful approach for refined epigenetic classification of brain tumors with heterogeneous histopathological features. Somatic mutations in the genes encoding isocitrate dehydrogenase (IDH; IDH1 and IDH2) are associated with improved prognosis [26, 73, 74] and have been observed in adolescent and younger adult patients with astrocytoma grades 2–4, or oligodendroglioma [75, 76]. IDH mutations induce a dramatic CpG island methylator phenotype (G-CIMP) [77]. This DNA hypermethylation signature is easily distinguished from other glioma subtypes and may contribute to differences in cellular plasticity between IDH WT and IDH mutant adult gliomas [78]. However, other glioma subgroups, even those arising in similar locations, can also be readily distinguished by genome-wide DNA methylation signatures. H3 G34-mutant diffuse hemispheric glioma clusters distinctly from IHG or H3- and IDH1-wildtype diffuse pediatric-type HGG arising in the cerebral hemispheres. IHG, meanwhile, bears a striking resemblance to low-grade gliomas when profiled in methylation arrays [22, 27]. H3 K27-altered DMG harboring K27M substitutions or other epigenetic alterations have distinguishing DNA methylation signatures and cluster separately from other pHGGs (Fig. 5a, b) [7, 72, 79, 80]. H3.1/2 K27M and H3.3 K27M DMG can be further subdivided by distinguishable chromatin states, gene expression profiles, and DNA methylation profiles [80], consistent with distinct developmental origins and/or biological processes driving growth in these tumor types, which may be further compounded by the specific oncohistone and associated secondary mutations.

DNA methylation classification of common childhood and adult brain tumors. tSNE projections of tumor methylation classes from published brain malignancy methylation datasets. a) tSNE projection of full reference cohort (n = 2751) with high grade gliomas encircled. b) tSNE projection of high-grade gliomas (n = 1259). Samples are shaped based on the study from which they originated [27, 70, 79]. The methylation classes are grouped by tumor type and color-coded (pHGG: pediatric high-grade glioma; GBM: glioblastoma multiforme; LGG: low grade glioma; EPN: ependymoma; ATRT: atypical teratoid rhabdoid tumors; MB: medulloblastoma). tSNE projections were generated from 1-variance weighted Pearson correlation between samples, which were calculated from normalized methylation beta values for the 35,000 most variable probes
3.2 Diffuse midline glioma, H3 K27-altered
Interestingly, a subset of DMGs were recently identified that lack the characteristic H3 K27M mutations, despite exhibiting DNA methylation patterns that closely resemble H3 K27M-mutant DMGs. To recognize that multiple mechanisms may contribute to gliomagenesis by depleting H3K27me3, the 2021 World Health Organization revision updated the subgroup nomenclature to “diffuse midline glioma, H3 K27-altered”. In posterior fossa subgroup A (PFA) ependymomas, overexpression, sometimes accompanied by mutation, of EZHIP (Ezh2-Interacting Protein, or CXorf67) drives a dramatic decrease in levels and spreading of H3K27me3, increases H3K27ac and is correlated with poorer survival as in H3 K27M-mutant gliomas [2, 81, 82]. Although ependymoma and DMG are distinct tumor entities, a rare subgroup of PFA ependymoma contain H3 K27M mutations instead of EZHIP overexpression, and rare DMG tumors with low H3K27me3 contain EZHIP overexpression rather than H3 K27M mutation [2, 83,84,85]. This suggests that different alterations leading to depletion of H3K27me3 contribute similarly to tumorigenesis.
A third subgroup of DMGs with H3K27me3 loss contain amplification, missense mutations, or small in-frame insertions of EGFR arising with unilateral or bilateral thalamic patterns, or in other midline locations. A subset of these DMGs contain H3 K27M mutation while others overexpress EZHIP. This subgroup contributes to the new classification of H3 K27-altered DMG and are distinguishable from an additional group of pediatric bithalamic gliomas with EGFR exon 20 in-frame insertions or exon 7 missense mutations that have distinct DNA methylation profiles [70, 86].
3.3 Cooperating mutations in pHGGs
In pediatric HGGs, hypermethylation of five CpG sites upstream of the TERT transcription start site has been linked with increased TERT expression, tumor progression, and poor survival [87, 88], likely driven by increased telomerase activity and telomere lengthening as in other cancers [89, 90]. ATRX mutations are mutually exclusive with TERT promoter mutations in adult gliomas and have frequently been observed in pHGGs [91, 92]. Such mutations provide an alternative method of telomere lengthening (ALT), which can prevent growth arrest in immortalized cells [93]. Together, these mutations indicate an important role for telomere maintenance during gliomagenesis and tumor growth.
The unique and highly subgroup-selective oncohistone mutations cooperate with more traditional cancer pathways to drive gliomagenesis. H3 K27 or G34 oncohistone mutations frequently co-occur with mutations found across glioma subgroups including mutations in the TP53, CyclinD/CKD4/6, receptor tyrosine kinase (RTK), and PI3-kinase (PI3K) pathways [3, 7, 17,18,19, 94]. However, there are several distinctive combinations that demonstrate selective cooperativity with specific oncohistone mutations and/or the developmental context of glioma subtypes. For example, H3.1 K27M frequently co-occurs with mutations in the bone morphogenetic protein (BMP) receptor ALK2 (ACVR1; 80%) and members of the PI3K signaling pathway, such as PIK3CA or PIK3R1 (~ 40%) (Fig. 6a) [7, 17,18,19, 94]. Some RTK mutations show regional selectivity, such as PDGFRA amplification in approximately 20–30% of brainstem H3.3 K27M DMGs, sometimes in association with activating point mutations [7, 17, 95, 96], while FGFR1 mutations are associated with thalamic H3.3 K27M DMG [7, 18]. MYC and MYCN amplifications have also been identified in H3.3 K27M DMGs, and both epigenetic activation of MYC and spontaneous MYC genomic amplification have been shown in a mouse model of H3.3 K27M-driven tumorigenesis and patient-derived H3.3 K27M cell lines [19, 97]. In contrast to H3 K27M DMG, H3 G34-mutant diffuse hemispheric glioma almost invariably exhibits concomitant loss of function mutations in TP53 and the histone H3.3 chaperone protein, ATRX [3, 7, 17, 69, 72]. Moreover, activating mutations in PDGFRA are found in almost half of all H3.3 G34-mutant diffuse gliomas at diagnosis. Intriguingly, this frequency increases to 81% in the setting of recurrent H3 G34-mutant tumors [21], indicating a strong selective pressure for PDGFRA activation in therapy-resistant tumor cells and a potential therapeutic target (Fig. 6C).

Cooperating mutations differ between oncohistone-driven tumors. Although many cooperating mutations are shared across oncohistone-driven tumors, some occur at much higher frequency in combination with specific oncohistones. Cooperating mutations with selective high frequency associations with histones a) H3.1/2 and b and c) H3.3 are denoted with asterisks. (TF: transcription factor)
4 Invasive phenotype of DMG, H3 K27M-altered
HGGs often exhibit intraparenchymal restriction, unlike other malignancies, and rarely present with extra-neural metastases [98, 99]. Rather, neoplastic cells infiltrate the perivascular space and brain parenchyma, becoming diffusely intertwined within the complex network of neuronal, glial, and vascular architecture [100]. In the most extreme manifestation of HGG infiltration, gliomatosis cerebri, tumor cells display a diffuse spreading pattern involving multiple brain regions. While histologically indistinguishable from adult tumors, pediatric HGGs harboring distinct histone mutations demonstrate clinicopathologic and radiologic features correlating with prognosis. For instance, H3K27-altered DMGs are uniformly aggressive entities irrespective of histology. Moreover, their anatomical location precludes surgical resection resulting in radiation therapy becoming the mainstay of care [101].
In contrast, H3 G34-mutant diffuse hemispheric gliomas are often amenable to maximally-safe gross total resection (GTR) by radiographic standards. This is particularly critical as the degree of surgical resection is the most important prognostic indicator of progression-free survival in children with supratentorial HGG independent of other prognostic factors. Nevertheless, in the context of diffusely infiltrative hemispheric HGGs, the presence of microscopic tumor cells interwoven throughout healthy brain tissue prohibits the most experienced neurosurgeons from removing all of the cancer cells [102]. While the standard of care for children with midline and hemispheric gliomas remains radiation therapy and maximally-safe GTR, respectively, the widespread tumor infiltration allows cancer cells to evade current treatment approaches contributing to significant morbidity and mortality.
The diffusely infiltrative nature of H3 K27-altered DMG has allowed for the evolutionary reconstruction of driver mutations through spatial genomic studies of DIPG autopsy samples. Multiple postmortem tumor samples per individual were collected from varying locations within the pons as well as in contiguous or distal extrapontine brain regions. Whole exome sequencing showed examples with evidence of distal tumor invasion to regions as far as the frontal lobe. In all cases, when H3 K27M mutation was present in any of the samples from an individual, it appeared to be a founding mutation, present in all locations with evidence of tumor, demonstrating an early clonal H3 K27M mutation. Secondary mutations in TP53, ACVR1, the PI3K pathway, ATRX or amplification of PDGFRA were recurrently detected, although these were more variable between tumor locations from an individual, consistent with subclonal populations and tumor evolution. Interestingly, there were multiple examples of convergent evolution, with distinct somatic variants in the catalytic or regulatory subunits of PI3K arising independently, revealing strong selective pressure for PI3K mutations later in tumor evolution [103,104,105,106]. 50% of tumor regions examined contained mixed subclones reflecting multiple lineages and waves of tumor invasion [103]. Isolation and analysis of single cell-derived subclones cultured alone or in combination indicated enhanced growth, migration, and invasion properties of mixed populations, suggesting a selective advantage for maintaining subclonal diversity [106]. Taken together, these studies support the conclusion that H3 K27M mutation is an early founding clonal event in DMG, with no other specific secondary mutation consistently required to drive extrapontine invasion. The highly invasive properties of these tumors pose a major therapeutic challenge and are consistent with the devastating prognosis for this disease.
5 Developmental origins of histone-mutant glioma
Many pediatric cancers are diseases of development gone awry due to dysregulation of developmental genes involved in stem cell maintenance, proliferation, differentiation, and cell fate [107, 108]. Mammalian brain development is a complex process occurring in a highly coordinated spatiotemporal manner. Neural stem cells divide to self-renew or produce cells that transition through fate or lineage-committed progenitor cell states and further differentiate into specialized mature neurons and glia in an elegant stepwise fashion to produce the right cell type with the right function in the right place and at the right time. This process is regulated through intricate crosstalk of extrinsic and intrinsic signaling. Chromatin structure and epigenetic regulation are highly involved in regulation of cellular states and differentiation. Spatial and single-cell omics methods have made great strides in uncovering regional and age-dependent epigenetic differences across distinct cell types [109,110,111], providing detailed illustrations of the role of epigenetic regulation during development.
Transcriptome comparisons of tumors and developing normal cells in the human and mouse brain suggest that oncohistones may transform precursor cells in specific developmental contexts [112]. The expression signatures of H3.1 and H3.3 K27M tumors, for example, mirror distinct stages of oligodendroglial maturation [113, 114] despite exhibiting oncogenic transformation potential in early pluripotent progenitors [32, 45, 51, 115]. In vitro models of forebrain or hindbrain human embryonic stem cell (hESC) spheroids and neural stem cells (NSC) showed differential effects of H3.3 K27M and H3.3 G34R mutations depending on the context of regional identity [64, 66]. H3.3 K27M increased proliferation and clonogenicity and reduced senescence selectively in human brainstem NSCs while H3.3 G34R did the opposite [64]. Overexpression of WT H3.3 and H3.3 G34R in combination with ATRX knockout and p53 loss in human induced ventral forebrain spheroids increased SOX2 + positive progenitor cells and proliferation but had no effect in ventral hindbrain spheroids [66]. These findings suggest that some developmentally-regulated epigenetic signatures become, in essence, ‘frozen’ in time upon oncohistone-driven oncogenic transformation. Tumors induced in mouse models from H3 K27M-expressing progenitor cells similarly retained epigenetic [45] and transcriptomic [112,113,114] signatures related to region-specific developmentally-regulated genes. In addition, DNA methylation patterns in human gliomas correlate strongly with epigenetic and transcriptomic profiles related to tumor location [79].
Most gliomas, including H3 K27-altered DMG, exhibit high expression of OLIG2, an essential transcription factor for the specification of oligodendrocyte precursor cells (OPCs) in the developing brain [116, 117]. The postnatal human pons undergoes significant volumetric growth within the first 7 years of life that is driven by an increase in proliferative OLIG2 + glial precursor cells[118]. The murine pons undergoes a similar phase of early postnatal growth, coinciding with a massive expansion of glial progenitors, not myelination or neurogenesis [119]. The childhood onset and glial-like signatures of H3 K27-altered DMG suggests that an early postnatal wave of gliogenesis may provide a window of susceptibility for oncogenic transformation of glial progenitor cells during childhood.
The cellular state from which K27M-altered DMG originates is hypothesized to be a neural stem/progenitor cell (NSPC) or OPC. Single-cell transcriptomic characterization of patient H3 K27M DIPG samples recapitulates an abnormal version of a developmental hierarchy, with the majority of DIPG cells expressing OPC-like transcriptional signatures, and other subpopulations of tumor cells with more differentiated expression signatures resembling either oligodendrocytes or astrocytes [120, 121]. Another single-cell multi-omics study identified distinct OPC-like states in DMGs arising in different brain regions. In pontine DMG, OPC-like cells were enriched for pre-OPC and HOX gene signatures while thalamic DMG OPC-like cells were enriched for OPC, committed OPC, and thalamic patterning signatures. Interestingly, a distinct boundary of HOXA8 was expressed in H3.1 but not H3.3 K27-altered DMGs, suggesting a unique and specific domain of origin for H3.1 K27-altered tumors. H3.1 K27-altered DMGs were also enriched for first-wave ventral sonic hedgehog (SHH)-specified NKX6.1 expression while H3.3 K27-altered DMGs were enriched for second-wave BMP/WNT-specified PAX3 [121]. Earlier studies identified Pax3 + progenitor cells, which co-expressed the neural stem and progenitor cell markers Nestin and Sox2, exclusively in brainstem and midline locations in the early postnatal mouse [122, 123]. H3.3 K27M expression combined with PDGFB overexpression and p53 loss in Pax3-expressing cells drives brainstem and midline gliomagenesis in mice, providing evidence that Pax3-expressing brainstem and midline progenitors are competent for tumorigenesis [123]. These data support the presence of distinct developmental origins of DMG likely determined by location, developmental state and histone variant context and suggest that oncohistone mutant gliomas carry molecular signatures that both reflect developmental origins and alter transcriptomic profiles to promote or sustain growth.
Despite the predominantly OPC-like signatures associated with H3 K27M DMG, various mouse models have shown that the introduction of H3 K27M with cooperating glioma mutations into Nestin + neural progenitor cells (NPCs) is sufficient to induce high-grade gliomas [45, 46, 115, 124, 125]. Indeed, H3 K27M can induce gliomagenesis when expressed in either Nestin + NPCs or Olig2 + OPCs [126]. Interestingly, a human induced pluripotent stem cell (iPSC)-derived model in which the H3.3 K27M mutation was engineered into the endogenous H3F3A allele suggests K27M, with TP53 knockdown, may confer tumorigenicity in the iNSC-state but not the-iOPC state, conflicting with the OPC origin hypothesis [51]. However, this in vitro model of neurodevelopment may lack important biological mechanisms related to cell-intrinsic regional identity that likely play a role in K27M-mediated tumorigenesis. These studies suggest that oncohistone mutations like H3 K27M may arise at early stages during cellular development but may only become growth permissive during transition states along the oligodendrocyte lineage. Significant intertumoral heterogeneity may also reflect variations in the precise cell state at transformation. Detailed understanding of the mechanistic connections between oncohistones and developmental origins of DMG will require further experimentation to elucidate the combinatorial effects of cellular states, unique origins, and developmental dynamics in brainstem and other midline structures.
With transcriptional signatures reminiscent of the developing forebrain, H3 G34-mutant diffuse hemispheric gliomas are characterized by a developmental program distinct from H3 K27-altered DMGs [69]. Analysis of a large multi-institutional cohort of patient tumors revealed differential enrichment for genes involved in the specification of interneuron lineage expression programs in H3 G34-mutant diffuse hemispheric gliomas [21]. Consistent with this, models of H3 G34R combined with cooperating mutations in human ES-cell induced or fetal progenitor cells showed the greatest oncogenic activity in cells with expression signatures resembling interneuron progenitor cells [64, 66], and expression of H3.3 G34R decreased proliferation and induced a senescence-like phenotype in brainstem NSCs [64], recapitulating a regional-specific selective advantage for H3 G34 mutations. These studies indicate that oncohistones may require the unique developmental contexts and genomic landscapes provided by specific progenitor cells to promote tumorigenesis (Fig. 7).

Cell states during development may be uniquely susceptible to oncohistone-driven transformation into tumors. Radial glial cells give rise to transit amplifying neural progenitor cells (NPCs) and glial progenitors. Neurogenesis driven by rapid NPC proliferation and differentiation into mature neurons precedes gliogenesis, where glial progenitors like intermediate glial cells (iGCs) and oligodendrocyte precursor cells (OPCs) differentiate into astrocyte or oligodendrocyte lineages respectively. The introduction of oncohistones at specific points in development, such as H3 G34R/V in interneuron progenitors or H3 K27M at different stages of early glial development, can cooperate with other acquired mutations to transform cells into tumors. Solid arrows show normal development. Dotted arrows show where specific mutations can lead to tumorigenesis
6 Oncohistones: Epigenetic dysregulation, disrupted development, and tumorigenesis in pHGG
6.1 H3 K27M alters differentiation
The incidence of oncohistone mutations in pediatric tumors suggests that these mutations may contribute to tumorigenesis by dysregulating cell differentiation and promoting proliferation during early development. H3 K27M alone has been shown to increase proliferation and self-renewal in mouse NSCs, hESC-derived NPCs, and iPSC-derived NSCs and OPCs [45, 51, 115], possibly through upregulation of PDGFRA [51]. Interestingly, H3 K27M reduces proliferation when expressed in hESC-derived astrocytes, primary human astrocytes, fibroblasts, or iPSCs [51, 115], suggesting pro-tumorigenic effects of H3 K27M may be permissive only in specific cellular states. In combination with PDGFRA overexpression and p53 loss, H3 K27M reduces oligodendrocyte differentiation and completely blocks astrocytic differentiation [115]. Interestingly, H3.3 K27M preferentially increases growth in brainstem NSCs [64] and results in focal epigenetic alterations at specific PRC1/2-bound neurodevelopmental genes [44].
Notably, depletion of H3 K27M in PDXs reduced proliferation and decreased neural and glial progenitor signatures and increased oligodendrocyte differentiation signatures [52], suggesting that H3 K27M may be promoting, restricting, and maintaining the OPC-like state while inhibiting differentiation. These experiments additionally showed that much of the transcriptomic variation between xenograft models remained after H3 K27M depletion, illustrating that inter-tumoral heterogeneity is likely influenced by developmental origins and contributing genetic mutations. Conversely, CRISPR-based knockout of H3 K27M in PDX enhanced astrocytic differentiation when subjected to astrocytic differentiation media conditions [112]. Disrupting the BAF chromatin remodeling complex revealed an epigenetic dependency for H3 K27M DMG and showed that BRG1 both maintains the OPC-like state and prevents tumor cell differentiation [127, 128]. Together, these studies indicate that promotion of progenitor state maintenance and inhibition of DMG cell differentiation may be essential oncogenic activities of H3 K27M.
6.2 H3 K27M in tumorigenesis
Although H3 K27M is an early or initiating event in DMG based on clonal mutations, no model thus far has generated tumors by introduction of H3 K27M as the sole driving event. This suggests that K27M may occur as an initiating event that provides a relatively small selective advantage while poising cells in a state where additional mutations will have a greater impact in inducing tumorigenesis. Alternatively, it is possible that existing model systems do not recapitulate the ideal cellular state for DMG initiation. Incorporation of H3 K27M-associated genetic mutations in model systems have been used to faithfully recreate and study the role of H3 K27M in gliomagenesis. H3.3 K27M cooperates with p53 loss and overexpression of PDGFRA, ATRX knockdown, or PDGFB overexpression to accelerate tumorigenesis when introduced exogenously into NPCs in vivo or in vitro [46, 115, 123, 124, 126, 129]. Conditional knock-in of H3.3 K27M combined with p53 knockout and PDGFRA activation in neonatal neural stem and progenitor cells selectively drove gliomagenesis at higher frequency in brainstem and hindbrain regions when compared to H3 WT tumors, displaying increased expression of genes associated with early neurodevelopmental signatures. Such models emphasize the region and age-selective effect of H3 K27M [45].
Similarly, H3.1 K27M, while not necessary for tumor growth, significantly accelerated tumorigenesis when combined with cooperating mutations Acvr1G328V and Pik3caH1047R in Olig2 + progenitors and may play a role in blocking differentiation [130]. H3 K27M depletion in H3K27-altered DMG xenografts reduced tumor growth and prevented cells from implanting into host tissue, illustrating a role or dependency for H3 K27M expression in tumor initiation and growth [42, 52, 54].
6.3 H3 G34R/V and oncogenesis
Although H3 G34 mutations lack the clear dominant epigenetic consequences associated with H3 K27M mutation, model systems have demonstrated some potential oncogenic contributions. Expression of cooperating mutations, ATRX and TP53, along with H3.3 G34R promoted proliferation and blocked differentiation in ventral forebrain progenitors, potentially through the expression of NOTCH2NL and suppression of intron retention [64, 66]. The importance of NOTCH signaling in H3 G34 and H3 K27-altered tumors was further corroborated using patient-derived cell lines edited with CRISPR-Cas9, where H3.3 K27M and H3.3 G34R induced genome-wide changes that induced expression of neurogenesis and NOTCH pathway genes to promote growth and survival [131]. A potential mechanism for reinforcing forebrain interneuron progenitor transcriptional circuitries is through a G34R-dependent disruption of H3.3 and ZMYND11 protein–protein interactions. ZMYND11 is a specific reader of H3.3 K36me3 and represses transcription by modulating the elongation and splicing of highly expressed genes. In H3 G34R-mutant patient-derived and engineered forebrain neural stem cell models, H3.3 K36me3 was preferentially lost at the promoters and genic regions of forebrain-associated genes with increased H3.3-G34R occupancy, possibly preventing ZMYND11-mediated repression and resulting in the maintenance of a forebrain progenitor state [64].
H3 G34-mutant gliomas are unusual among gliomas because expression of OLIG2 is largely absent [4]. However, PDGFRA, a regulator of proliferation and maintenance of the OPC-state, is expressed and frequently activated by mutation in these tumors (Fig. 2C). H3 G34-mutant glioma also exhibits aberrant H3K27ac deposition at the PDGFRA promoter and at a cis-regulatory element adjacent to the interneuron defining gene, GSX2. Enhancer hijacking results in H3.3 G34-mutant interneuron progenitors commandeering PDGFRA expression [21], which may contribute to the oncogenic potential of H3 G34 mutations. PDGFRA has been shown to be a dependency in glioblastoma and DIPG models [95, 120] and additionally appears to be a shared oncogenic mechanism in H3 G34-mutated glioma.
7 Therapeutic vulnerabilities of oncohistone mutant glioma
The discovery of oncohistone mutations in diffuse glioma raised the exciting possibility that these incurable brain tumors may have previously unexplored and potent sensitivities to drugs related to epigenetic reprogramming. The development of in vitro and in vivo models that faithfully represent the developmental context and mutation spectrum that uniquely define oncohistone-driven glioma has been crucial to understanding epigenetic dysregulation in these tumors and investigating more effective therapies for patients. Concerted efforts from multiple labs produced patient-derived tumor cell lines that recapitulate key genomic and epigenomic features of patient tumors and can be grown adherently in serum-free media or as neurospheres [106, 132,133,134]. Patient-derived orthotopic xenograft models grown in immunodeficient mouse hosts provide critical human tumor models for in vivo preclinical testing of brain tumor therapies, and allograft models of mouse tumor cells engineered to express hallmark mutations of histone mutant glioma provide useful models to further interrogate therapeutic response of tumor cells within an intact and complex inflammatory microenvironment [132, 133, 135]. Together, these in vitro and in vivo model systems provide unique and complementary avenues to uncover the mechanisms underlying oncohistone-driven tumor growth and discover new therapeutic vulnerabilities.
Epigenetic writers and erasers have been explored as therapeutic targets for oncohistone mutant tumors. High-throughput in vitro screening of H3 K27M DMG cell lines identified histone deacetylase inhibitors (HDACi) as promising candidates for the treatment of H3 K27-altered DMG [136, 137]. This sensitivity may relate, in part, to the aberrant increase in H3K27ac and associated upregulation of ERV expression observed in H3 K27M DMG [54]. However, a phase I trial testing dose-limiting toxicities of the HDACi panobinostat following radiation did not show significant improvement in progression-free or overall survival within the cohort enrolled on the phase I trial [138]. Combination therapies may be required to achieve greater efficacy. Indeed, combination of HDAC inhibition with lysine-specific demethylase 1 (LSD1) inhibition or simultaneous HDAC and proteosome inhibition can produce a synergistic effect to more effectively block tumor growth in vitro and in vivo [139]. Interestingly, combined HDAC and PI3K inhibition can also induce a DNA damage response that sensitizes pHGGs to radiation [140]. Expression of H3.1 and H3.3 K27M mutations sensitize cells to in vitro replication stress and may cause genomic instability due to impaired non-homologous end-join (NHEJ) and double-stranded break (DSB) repair [141], providing another potential vulnerability.
Targeting the polycomb repressive complex 1 (PRC1) protein BMI1, which may be upregulated by H3 K27M, similarly reduced tumor growth in xenograft models [142, 143]. Therapeutic targeting of EZH2 to deplete residual PRC2 activity in H3 K27M mutant glioma inhibited tumor cell growth in a mouse H3 K27M glioma model and in patient-derived DMG cell lines [35, 46]. The consequence of EZH2 inhibition may vary significantly, as it failed to inhibit tumor cell growth of some human pediatric DMG and HGG cells with or without H3 K27M mutation, and genetic deletion of Ezh2 in a mouse DMG model with wild-type H3 actually increased tumor grade and proliferation rates [144, 145]. In addition to limited success in treating DMG cells by targeting epigenetic writers and erasers directly, targeting downstream aberrant transcriptional activation through inhibition of CDK7 or CDK9, which regulate transcriptional initiation or elongation, respectively, induced cell death and inhibited proliferation of DMG cells [57, 132]. Combination of BET bromodomain protein inhibitors with panobinostat or inhibitors targeting other epigenetic writers such as CBP demonstrated enhanced cytotoxicity compared to single agent use [35, 146]. Targeting aberrant transcriptional activation through inhibition of HDACs, BRDs, and CDK7 or CDK9 is not only a promising therapeutic avenue but a useful tool to study the H3 K27M oncogenic programs contributing to DMG [35, 57, 132].
Metabolic studies showed that H3.3 K27M induced increased glycolysis, glutaminolysis, and tricarboxylic acid cycle metabolism leading to elevated levels of alpha-ketoglutarate that contributed to maintaining the low H3K27me3 levels associated with H3.3 K7M. Disrupting this interplay between epigenetic and metabolic dysregulation by inhibiting these metabolic pathways created a therapeutic vulnerability and inhibited growth of H3.3 K27M tumor cells [147]. DMG is also dependent on adequate levels of nicotinamide adenine dinucleotide (NAD +) and is sensitive to NAMPT inhibition [132, 137, 148]. Other metabolic vulnerabilities of pHGG include tumor cell death induced by redirecting glucose metabolism from glycolysis to mitochondrial oxidation through treatment with AICAR (5-aminoimidazole-4-carboxamide ribonucleotide) or the pyruvate dehydrogenase kinase inhibitor dichloroacetate (DCA) [149].
Efforts are underway to directly target novel neoantigens generated by the H3.3 K27M mutation through T-cell therapy or targeted vaccines [150, 151]. Additional approaches that employ immunotherapy, although they do not directly target the oncohistone mutation, are showing exciting promise. Adoptive T-cell therapies have risen to the forefront as a novel approach to leverage immune cells to target antigens on the surface of tumor cells. Preclinical studies using engineered CAR T-cells to target GD2-expressing tumor cells have demonstrated strong anti-tumorigenic potential [152]. Despite causing neuroinflammation and edema in preclinical xenograft models and in some patients treated with CAR T-cell therapy, the preliminary results from ongoing clinical trials remain promising, with all 4 reported DMG patients exhibiting radiographic and clinical improvement [153]. B7-H3 has also been identified as a promising target for CAR T-cell therapy in preclinical studies with H3 K27M DMG as well as H3 G34R mutant and H3 WT pediatric diffuse glioma and other pediatric brain tumors [154]. Recent studies have highlighted how tumor cells cooperate with the surrounding microenvironment to promote tumor growth [155,156,157]. EZH2 inhibition in microglia, for example, has been shown to decrease tumor growth and migration in H3 K27M patient-derived pHGG cell lines, indicating a potential for growth-promoting crosstalk between immune cells and tumors within the tumor microenvironment [158].
Earlier discovery of dopamine receptor signaling as a potential therapeutic target for adult glioblastoma [159, 160] led to clinical trials for glioma. ONC201, a postulated DRD2 antagonist, reduced H3 K27M-mutant DMG tumor growth [161] and demonstrated efficacy against H3 K27M-mutant tumors in clinical trials [162]. While these preliminary findings are exciting, the mechanism of action for ONC201 in H3 K7M mutant DMG therapy remains unclear. ONC201 has been described as a TNF-related apoptosis-inducing ligand (TRAIL)-inducing compound, DRD2 inhibitor, and, in some studies, an inhibitor of AKT/ERK signaling. ONC201 is also an allosteric agonist of the mitochondrial protease caseinolytic protease P (ClpP) [161, 163,164,165]. ONC201 and related compounds, their mechanisms of action, and sources of selectivity and resistance are active areas of investigation in ongoing clinical trials and laboratory studies by multiple investigators.
Novel therapeutic approaches for H3.3 G34 mutant glioma have been much less extensively studied relative to H3 K27M DMG. Epigenetic analyses of H3.3 G34R glioma cells showed redistribution of H3K27me3 to intergenic regions associated with enhanced leukemia inhibitory factor (LIF) expression. Elevated LIF drove an autocrine/paracrine stimulation of STAT3 activation, and STAT3 inhibition suppressed growth of H3.3 G34 diffuse glioma in vitro and in vivo [67].
High-throughput and focused screening studies showed encouraging consistency for many of the targets discussed above and a larger collection that have yet to be explored individually in detailed studies [57, 132, 136, 137]. It is noteworthy that despite the profound and unique consequence of the H3 K27M mutation on the epigenome, in a high-throughput screening study that included H3 K27, H3 G34R, and H3 WT pHGG cell lines, the oncohistone mutations did not confer unique vulnerabilities. Potent drugs displayed effects across all pHGG, and variability associated as much with intertumoral heterogeneity as with oncohistone mutation status [132]. Despite this, the encouraging preliminary results in this realm are driving new mechanistic studies, focused preclinical studies to identify synergistic therapeutic combinations, and the development of new clinical trials.
8 Additional histone mutations in diverse tumor types
In addition to the highly recurrent oncohistone mutations found in H3 K27M-altered DMG and H3 G34-mutant diffuse hemispheric glioma, a growing number of related and novel histone mutations have been identified in diverse tumor types.
Much like the disruption of H3K36 methylation status exhibited in H3 G34-mutant diffuse hemispheric gliomas, complementary mechanisms are employed as oncogenic drivers in other forms of cancer. Glycine to tryptophan/leucine substitutions at amino acid 34 in histone H3.3 (H3 G34W/L) molecularly define giant cell tumors of bone, where they occur with 92% frequency, but are rarely observed in osteosarcoma [166]. Unlike H3 G34R/V and H3 G34W/L substitutions which occur near the K36 amino acid, missense mutations have also been found directly at H3K36 in other malignancies. Lysine to methionine/isoleucine substitutions at amino acid 36 in histone H3 (H3 K36M/I) occur in 95% chondroblastoma and less commonly in histiocytic tumors of the skull, infantile soft tissue sarcomas, melanoma, head and neck squamous cell carcinomas, colorectal, and bladder cancer [166,167,168].
Given the importance of chromatin remodeling on cellular state, it is not surprising that H3 K36M/I-mediated epigenomic reprogramming disrupts downstream biological processes important for oncogenesis. In contrast to H3 G34-mutations that confer a loss of H3K36me3 only on those histones harboring the substitution, H3 K36M/I mutations decrease the global abundance of H3K36me1/2/3 across the epigenome analogous to the dominant-negative effect exerted by H3 K27M [169, 170]. In response, H3K27me3 levels are reciprocally increased at intergenic regions promoting the recruitment of PRC1 away from gene-associated H3K27me3. This leads to the aberrant restriction of chromatin architecture, thereby blocking the expression of genes important for cellular differentiation [169]. For example, in mouse allograft models of H3 K36-mutant sarcoma-like tumors, the atypical recruitment of PRC1 to intergenic regions disrupts transcriptional programs associated with mesenchymal progenitor cell differentiation to promote sarcomagenesis [169, 170].
Very rare H3.3 mutations encoded by H3F3B K27I substitutions that also cause global loss of H3K27me3 were found in DMG [171]. Subclonal H3 K27M/I mutations are also rarely observed in acute myeloid leukemia. Studies investigating the oncogenic mechanisms of H3 K27M in the context of leukemia show that H3 K27M increases proliferation, expansion, and self-renewal of hematopoietic stem and progenitor cells and inhibits differentiation. Interestingly, H3 K27M accelerates leukemia progression in an AML model but not MLL [172, 173], illustrating a context-dependent advantage similar to the cell-type specific effects of H3 K27M in neural cells.
In addition to recurrent hotspot mutations in the N-terminal tail of histone H3, lower frequency mutations have been identified in the globular folding domains of all four core histone families [174, 175]. Although a small number of residues within the globular domain of histones undergo post-translational modification, mutations at these sites do not alter epigenetic marks. Instead, these mutations disrupt the histone-histone interface and destabilize nucleosome structure. This structural disturbance alters histone dimer exchange dynamics, nucleosome sliding, and chromatin remodeling processes [176]. Unlike H3 K27, H3 G34, and H3 K36 mutations which disrupt the deposition and maintenance of post-translational marks associated with transcriptional regulation, the correlation between mutational status and downstream effects on transcription is less clear for mutations within globular domains. While there is a strong association of H3K27ac, H3K36me3, and H3K27me3 with epigenetic-mediated transcriptional control, predicting the impact of destabilized nucleosome structure on the transcriptome represents an emerging challenge.
Importantly, histone globular domain mutations have been shown to inhibit cellular differentiation by altering the expression of lineage-defining transcription factors and increasing chromatin accessibility at the promoter of a known developmentally associated gene, HOXB6 [174, 176]. Despite blocking differentiation in murine mesenchymal progenitor cells, these mutations do not exhibit clear selection pressures for a particular developmental context or specific cellular state based on their mutational patterns across human cancer types. Unlike the different H3 mutations which occur at a high frequency and show striking specificity for subgroups of diffuse glioma, chondroblastoma, and giant cell tumors of bone in children and adolescents, globular domain mutations occur in a variety of adult cancers. Indeed, understanding why mutations in globular domains exhibit less tumor type specificity compared to conventional oncohistones (H3 K27, H3 G34, and H3 K36) may yield important insights. With somatic mutations in histones occurring in approximately 4% of diverse tumor types spanning a developmental continuum, understanding the broader role of these mutations in oncogenesis will undoubtedly inform epigenetic-based cancer therapies and provide a deeper understanding of the unique developmental associations with certain alterations [175].
9 Concluding remarks
From the discovery of a DNA hypomethylation phenotype in carcinoma in the early 1980s [177] to the more recent identification of oncohistones in pediatric HGGs in the last decade [3, 5], epigenetic reprogramming has now been recognized as one of the hallmarks of cancer [178]. Histone H3 mutations disrupt the strict spatial and temporal control of specific developmental cellular states by modifying chromatin structure and causing widespread gene expression changes. Oncohistones generate unique epigenetic and transcriptomic landscapes, which cooperate with other acquired mutations to promote tumorigenesis under specific contexts. Analyses of these mutations and the tumors in which they arise led to a significant change in the way that brain tumors are classified worldwide. Functional demonstrations of the requirement for high-frequency oncohistone mutations in tumor maintenance also support the growing evidence that low-frequency mutations in histones across multiple tumor types may contribute to tumorigenesis.
Despite the dramatic progress in understanding consequences of oncohistones on the epigenome and transcriptome, many unanswered questions regarding the mechanisms driving oncohistone effects in cancer remain. Fundamental effects of H3 K27M and H3 G34R/V mutations on the levels and spreading of H3K27me2/me3 and H3K36me2/me3 connect the two mutations with each inducing different shifts in the balance of these two posttranslational marks. However, selective changes in gene expression downstream of these mutations are still incompletely understood. Genes with bivalent promoters are more susceptible to expression changes induced by H3 K27M mutations, but what determines which specific genes are most dramatically impacted? Are other expression changes indirect effects, or selectively regulated by mechanisms not yet understood? H3 G34R/V mutations are less well understood, without the clear dominant impact on the epigenome exerted by H3 K27M. How might oncohistone mutations impact other aspects of genome dynamics outside of transcription that are important in cancer such as replication, telomere maintenance and DNA repair?
The spatiotemporal incidence of oncohistone mutations shows an intriguing selective advantage connected to unique developmental states and oncohistone-regulated genes are highly enriched for those impacting neurodevelopmental processes. The use of current and emerging approaches to evaluate the epigenome at the single-cell level to probe questions related to intratumoral heterogeneity and oncohistone deposition, chromatin dysregulation, and tumor cell states may yield new insights into the contribution of oncohistones to cell state as well therapeutic resistance.
Studying rare tumor types may have much broader implications for understanding epigenetic misregulation as a driving force in cancer, particularly in common cancer types in which only rare histone mutations are observed. What proportion of the changes induced by oncohistones are essential contributors to their oncogenic activity? Most importantly, how can histone misregulation be leveraged as a potent therapeutic vulnerability for cancers with and without oncohistone mutation? While detailed molecular studies have highlighted likely developmental origins for histone mutant glioma, it remains unclear why these contexts constitute uniquely permissive or selective conditions for oncohistone-mediated transformation. A complete understanding of the developmental regulation of epigenetic states and how oncohistones co-opt these states to permit tumorigenesis and tumor growth will be instrumental to determine precision therapeutic approaches and improve outcomes for patients with these incurable diseases.
References
Ostrom, Q.T., Price, M., Ryan, K., Edelson, J., Neff, C., Cioffi, G., et al. (2022). CBTRUS statistical report: pediatric brain tumor foundation childhood and adolescent primary brain and other central nervous system tumors diagnosed in the United States in 2014–2018. NLM (Medline), iii1–iii38. https://doi.org/10.1093/neuonc/noac161
Pajtler, K. W., Wen, J., Sill, M., Lin, T., Orisme, W., Tang, B., et al. (2018). Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathologica, 136(2), 211–226. https://doi.org/10.1007/s00401-018-1877-0
Schwartzentruber, J., Korshunov, A., Liu, X. Y., Jones, D. T., Pfaff, E., Jacob, K., et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature, 482(7384), 226–31. https://doi.org/10.1038/nature10833
Sturm, D., Witt, H., Hovestadt, V., Khuong-Quang, D. A., Jones, D. T., Konermann, C., et al. (2012). Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell, 22(4), 425–437. https://doi.org/10.1016/j.ccr.2012.08.024
Wu, G., Broniscer, A., McEachron, T. A., Lu, C., Paugh, B. S., Becksfort, J., et al. (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature Genetics, 44(3), 251–253. https://doi.org/10.1038/ng.1102
Ceccarelli, M., Barthel, F. P., Malta, T. M., Sabedot, T. S., Salama, S. R., Murray, B. A., et al. (2016). Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell, 164(3), 550–563. https://doi.org/10.1016/j.cell.2015.12.028
Mackay, A., Burford, A., Carvalho, D., Izquierdo, E., Fazal-Salom, J., Taylor, K. R., et al. (2017). Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell, 32(4), 520-537 e5. https://doi.org/10.1016/j.ccell.2017.08.017
Mendiratta, S., Gatto, A., & Almouzni, G. (2019). Histone supply: Multitiered regulation ensures chromatin dynamics throughout the cell cycle. Journal of Cell Biology, 218(1), 39–54. https://doi.org/10.1083/jcb.201807179
Talbert, P.B. and Henikoff, S. (2021). Histone variants at a glance. Journal of Cell Science, 134(6). https://doi.org/10.1242/jcs.244749
Filipescu, D., Muller, S., & Almouzni, G. (2014). Histone H3 variants and their chaperones during development and disease: Contributing to epigenetic control. Annual Review of Cell and Developmental Biology, 30, 615–646. https://doi.org/10.1146/annurev-cellbio-100913-013311
Lewis, P.W., Elsaesser, S.J., Noh, K.M., Stadler, S.C., and Allis, C.D. (2010). Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proceedings of the National Academy of Sciences, 107(32):14075–80. https://doi.org/10.1073/pnas.1008850107
Amorim, J.P., Santos, G., Vinagre, J., and Soares, P. (2016). The Role of ATRX in the Alternative Lengthening of Telomeres (ALT) Phenotype. Genes (Basel), 7(9). https://doi.org/10.3390/genes7090066
Lovejoy, C. A., Li, W., Reisenweber, S., Thongthip, S., Bruno, J., de Lange, T., et al. (2012). Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet, 8(7), e1002772. https://doi.org/10.1371/journal.pgen.1002772
Ray-Gallet, D. and Almouzni, G. (2019). Histone Mutations and Cancer. 17–42
Khuong-Quang, D. A., Buczkowicz, P., Rakopoulos, P., Liu, X. Y., Fontebasso, A. M., Bouffet, E., et al. (2012). K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathological, 124(3), 439–47. https://doi.org/10.1007/s00401-012-0998-0
Ren, M., & Van Nocker, S. (2016). In silico analysis of histone H3 gene expression during human brain development (pp. 167–173). University of the Basque Country Press.
Wu, G., Diaz, A. K., Paugh, B. S., Rankin, S. L., Ju, B., Li, Y., et al. (2014). The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nature Genetics, 46(5), 444–450. https://doi.org/10.1038/ng.2938
Fontebasso, A. M., Papillon-Cavanagh, S., Schwartzentruber, J., Nikbakht, H., Gerges, N., Fiset, P. O., et al. (2014). Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nature Genetics, 46(5), 462–466. https://doi.org/10.1038/ng.2950
Buczkowicz, P., Hoeman, C., Rakopoulos, P., Pajovic, S., Letourneau, L., Dzamba, M., et al. (2014). Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nature Genetics, 46(5), 451–456. https://doi.org/10.1038/ng.2936
Karremann, M., Gielen, G. H., Hoffmann, M., Wiese, M., Colditz, N., Warmuth-Metz, M., et al. (2018). Diffuse high-grade gliomas with H3 K27M mutations carry a dismal prognosis independent of tumor location. Neuro-Oncology, 20(1), 123–131. https://doi.org/10.1093/neuonc/nox149
Chen, C. C. L., Deshmukh, S., Jessa, S., Hadjadj, D., Lisi, V., Andrade, A. F., et al. (2020). Histone H3.3G34-Mutant Interneuron Progenitors Co-opt PDGFRA for Gliomagenesis. Cell, 183(6), 1617–1633. https://doi.org/10.1016/j.cell.2020.11.012
Guerreiro Stucklin, A. S., Ryall, S., Fukuoka, K., Zapotocky, M., Lassaletta, A., Li, C., et al. (2019). Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nature Communications, 10(1), 4343. https://doi.org/10.1038/s41467-019-12187-5
Korshunov, A., Ryzhova, M., Hovestadt, V., Bender, S., Sturm, D., Capper, D., et al. (2015). Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathologica, 129(5), 669–678. https://doi.org/10.1007/s00401-015-1405-4
Mackay, A., Burford, A., Molinari, V., Jones, D. T. W., Izquierdo, E., Brouwer-Visser, J., et al. (2018). Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell, 33(5), 829-842 e5. https://doi.org/10.1016/j.ccell.2018.04.004
Korshunov, A., Capper, D., Reuss, D., Schrimpf, D., Ryzhova, M., Hovestadt, V., et al. (2016). Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathologica, 131(1), 137–146. https://doi.org/10.1007/s00401-015-1493-1
Yan, H., Parsons, D. W., Jin, G., McLendon, R., Rasheed, B. A., Yuan, W., et al. (2009). IDH1 and IDH2 mutations in gliomas. New England Journal of Medicine, 360(8), 765–773. https://doi.org/10.1056/NEJMoa0808710
Clarke, M., Mackay, A., Ismer, B., Pickles, J. C., Tatevossian, R. G., Newman, S., et al. (2020). Infant High-Grade Gliomas Comprise Multiple Subgroups Characterized by Novel Targetable Gene Fusions and Favorable Outcomes. Cancer Discovery, 10(7), 942–963. https://doi.org/10.1158/2159-8290.CD-19-1030
Korshunov, A., Schrimpf, D., Ryzhova, M., Sturm, D., Chavez, L., Hovestadt, V., et al. (2017). H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathologica, 134(3), 507–516. https://doi.org/10.1007/s00401-017-1710-1
Bender, S., Tang, Y., Lindroth, A. M., Hovestadt, V., Jones, D. T., Kool, M., et al. (2013). Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell, 24(5), 660–672. https://doi.org/10.1016/j.ccr.2013.10.006
Chan, K. M., Fang, D., Gan, H., Hashizume, R., Yu, C., Schroeder, M., et al. (2013). The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes & Development, 27(9), 985–90. https://doi.org/10.1101/gad.217778.113
Venneti, S., Garimella, M. T., Sullivan, L. M., Martinez, D., Huse, J. T., Heguy, A., et al. (2013). Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathology, 23(5), 558–564. https://doi.org/10.1111/bpa.12042
Lewis, P. W., Muller, M. M., Koletsky, M. S., Cordero, F., Lin, S., Banaszynski, L. A., et al. (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science, 340(6134), 857–861. https://doi.org/10.1126/science.1232245
Diehl, K.L., Ge, E.J., Weinberg, D.N., Jani, K.S., Allis, C.D., and Muir, T.W. (2019). PRC2 engages a bivalent H3K27M-H3K27me3 dinucleosome inhibitor. Proceedings of the National Academy of Sciences, 116(44), 22152-22157https://doi.org/10.1073/pnas.1911775116
Fang, D., Gan, H., Cheng, L., Lee, J.H., Zhou, H., Sarkaria, J.N., et al. (2018). H3.3K27M mutant proteins reprogram epigenome by sequestering the PRC2 complex to poised enhancers. Elife, 7. https://doi.org/10.7554/eLife.36696
Piunti, A., Hashizume, R., Morgan, M. A., Bartom, E. T., Horbinski, C. M., Marshall, S. A., et al. (2017). Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nature Medicine, 23(4), 493–500. https://doi.org/10.1038/nm.4296
Joshi, A., Miller, C., Jr., Baker, S. J., & Ellenson, L. H. (2015). Activated mutant p110alpha causes endometrial carcinoma in the setting of biallelic Pten deletion. American Journal of Pathology, 185(4), 1104–1113. https://doi.org/10.1016/j.ajpath.2014.12.019
Justin, N., Zhang, Y., Tarricone, C., Martin, S. R., Chen, S., Underwood, E., et al. (2016). Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nature Communications, 7, 11316. https://doi.org/10.1038/ncomms11316
Stafford, J. M., Lee, C. H., Voigt, P., Descostes, N., Saldana-Meyer, R., Yu, J. R., et al. (2018). Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Science Advances, 4(10), eaau5935. https://doi.org/10.1126/sciadv.aau5935
Lee, C. H., Yu, J. R., Granat, J., Saldana-Meyer, R., Andrade, J., LeRoy, G., et al. (2019). Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma. Genes & Development, 33(19–20), 1428–1440. https://doi.org/10.1101/gad.328773.119
Sarthy, J.F., Meers, M.P., Janssens, D.H., Henikoff, J.G., Feldman, H., Paddison, P.J., et al. (2020). Histone deposition pathways determine the chromatin landscapes of H3.1 and H3.3 K27M oncohistones. Elife, 9. https://doi.org/10.7554/eLife.61090
Wang, X., Long, Y., Paucek, R. D., Gooding, A. R., Lee, T., Burdorf, R. M., et al. (2019). Regulation of histone methylation by automethylation of PRC2. Genes & Development, 33(19–20), 1416–1427. https://doi.org/10.1101/gad.328849.119
Harutyunyan, A. S., Krug, B., Chen, H., Papillon-Cavanagh, S., Zeinieh, M., De Jay, N., et al. (2019). H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nature Communications, 10(1), 1262. https://doi.org/10.1038/s41467-019-09140-x
Zhang, J., Ding, L., Holmfeldt, L., Wu, G., Heatley, S. L., Payne-Turner, D., et al. (2012). The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature, 481(7380), 157–163. https://doi.org/10.1038/nature10725
Brien, G. L., Bressan, R. B., Monger, C., Gannon, D., Lagan, E., Doherty, A. M., et al. (2021). Simultaneous disruption of PRC2 and enhancer function underlies histone H3.3–K27M oncogenic activity in human hindbrain neural stem cells. Nature Genetics, 53(8), 1221–1232. https://doi.org/10.1038/s41588-021-00897-w
Larson, J. D., Kasper, L. H., Paugh, B. S., Jin, H., Wu, G., Kwon, C. H., et al. (2019). Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell, 35(1), 140-155 e7. https://doi.org/10.1016/j.ccell.2018.11.015
Mohammad, F., Weissmann, S., Leblanc, B., Pandey, D. P., Hojfeldt, J. W., Comet, I., et al. (2017). EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nature Medicine, 23(4), 483–492. https://doi.org/10.1038/nm.4293
Wagner, E. J., & Carpenter, P. B. (2012). Understanding the language of Lys36 methylation at histone H3. Nature Reviews Molecular Cell Biology, 13(2), 115–126. https://doi.org/10.1038/nrm3274
Schmitges, F. W., Prusty, A. B., Faty, M., Stutzer, A., Lingaraju, G. M., Aiwazian, J., et al. (2011). Histone methylation by PRC2 is inhibited by active chromatin marks. Molecular Cell, 42(3), 330–341. https://doi.org/10.1016/j.molcel.2011.03.025
Yuan, W., Xu, M., Huang, C., Liu, N., Chen, S., & Zhu, B. (2011). H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. Journal of Biological Chemistry, 286(10), 7983–7989. https://doi.org/10.1074/jbc.M110.194027
Harutyunyan, A. S., Chen, H., Lu, T., Horth, C., Nikbakht, H., Krug, B., et al. (2020). H3K27M in Gliomas Causes a One-Step Decrease in H3K27 Methylation and Reduced Spreading within the Constraints of H3K36 Methylation. Cell Reports, 33(7), 108390. https://doi.org/10.1016/j.celrep.2020.108390
Haag, D., Mack, N., Goncalves, Benites, da Silva, P., Statz, B., Clark, J., Tanabe, K., et al. (2021). H3.3–K27M drives neural stem cell-specific gliomagenesis in a human iPSC-derived model. Cancer Cell, 39(3), 407-422 e13. https://doi.org/10.1016/j.ccell.2021.01.005
Silveira, A. B., Kasper, L. H., Fan, Y., Jin, H., Wu, G., Shaw, T. I., et al. (2019). H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathologica, 137(4), 637–655. https://doi.org/10.1007/s00401-019-01975-4
Furth, N., Algranati, D., Dassa, B., Beresh, O., Fedyuk, V., Morris, N., et al. (2022). H3–K27M-mutant nucleosomes interact with MLL1 to shape the glioma epigenetic landscape. Cell Reports, 39(7), 110836. https://doi.org/10.1016/j.celrep.2022.110836
Krug, B., De Jay, N., Harutyunyan, A. S., Deshmukh, S., Marchione, D. M., Guilhamon, P., et al. (2019). Pervasive H3K27 Acetylation Leads to ERV Expression and a Therapeutic Vulnerability in H3K27M Gliomas. Cancer Cell, 35(5), 782-797 e8. https://doi.org/10.1016/j.ccell.2019.04.004
Liu, M., Thomas, S. L., DeWitt, A. K., Zhou, W., Madaj, Z. B., Ohtani, H., et al. (2018). Dual Inhibition of DNA and Histone Methyltransferases Increases Viral Mimicry in Ovarian Cancer Cells. Cancer Research, 78(20), 5754–5766. https://doi.org/10.1158/0008-5472.CAN-17-3953
Hnisz, D., Abraham, B. J., Lee, T. I., Lau, A., Saint-Andre, V., Sigova, A. A., et al. (2013). Super-enhancers in the control of cell identity and disease. Cell, 155(4), 934–947. https://doi.org/10.1016/j.cell.2013.09.053
Nagaraja, S., Vitanza, N. A., Woo, P. J., Taylor, K. R., Liu, F., Zhang, L., et al. (2017). Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell, 31(5), 635-652 e6. https://doi.org/10.1016/j.ccell.2017.03.011
Nagaraja, S., Quezada, M. A., Gillespie, S. M., Arzt, M., Lennon, J. J., Woo, P. J., et al. (2019). Histone Variant and Cell Context Determine H3K27M Reprogramming of the Enhancer Landscape and Oncogenic State. Molecular Cell, 76(6), 965-980 e12. https://doi.org/10.1016/j.molcel.2019.08.030
Lowe, B.R., Yadav, R.K., Henry, R.A., Schreiner, P., Matsuda, A., Fernandez, A.G., et al. (2021). Surprising phenotypic diversity of cancer-associated mutations of Gly 34 in the histone H3 tail.Elife, 10. https://doi.org/10.7554/eLife.65369
Yang, S., Zheng, X., Lu, C., Li, G. M., Allis, C. D., & Li, H. (2016). Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes & Development, 30(14), 1611–1616. https://doi.org/10.1101/gad.284323.116
Brennan, C. W., Verhaak, R. G., McKenna, A., Campos, B., Noushmehr, H., Salama, S. R., et al. (2013). The somatic genomic landscape of glioblastoma. Cell, 155(2), 462–477. https://doi.org/10.1016/j.cell.2013.09.034
Fontebasso, A. M., Schwartzentruber, J., Khuong-Quang, D. A., Liu, X. Y., Sturm, D., Korshunov, A., et al. (2013). Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathologica, 125(5), 659–669. https://doi.org/10.1007/s00401-013-1095-8
Jain, S.U., Khazaei, S., Marchione, D.M., Lundgren, S.M., Wang, X., Weinberg, D.N., et al. (2020). Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proceedings of the National Academy of Sciences, 117(44), 27354–27364. https://doi.org/10.1073/pnas.2006076117
Bressan, R. B., Southgate, B., Ferguson, K. M., Blin, C., Grant, V., Alfazema, N., et al. (2021). Regional identity of human neural stem cells determines oncogenic responses to histone H3.3 mutants. Cell Stem Cell, 28(5), 877-893 e9. https://doi.org/10.1016/j.stem.2021.01.016
Fang, J., Huang, Y., Mao, G., Yang, S., Rennert, G., Gu, L., et al. (2018). Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSalpha interaction. Proceedings of the National Academy of Sciences, 115(38), 9598-9603https://doi.org/10.1073/pnas.1806355115
Funato, K., Smith, R. C., Saito, Y., & Tabar, V. (2021). Dissecting the impact of regional identity and the oncogenic role of human-specific NOTCH2NL in an hESC model of H3.3G34R-mutant glioma. Cell Stem Cell, 28(5), 894-905 e7. https://doi.org/10.1016/j.stem.2021.02.003
Sweha, S. R., Chung, C., Natarajan, S. K., Panwalkar, P., Pun, M., Ghali, A., et al. (2021). Epigenetically defined therapeutic targeting in H3.3G34R/V high-grade gliomas. Science Translational Medicine, 13(615), eabf7860. https://doi.org/10.1126/scitranslmed.abf7860
Voon, H. P. J., Udugama, M., Lin, W., Hii, L., Law, R. H. P., Steer, D. L., et al. (2018). Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma. Nature Communications, 9(1), 3142. https://doi.org/10.1038/s41467-018-05607-5
Bjerke, L., Mackay, A., Nandhabalan, M., Burford, A., Jury, A., Popov, S., et al. (2013). Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discovery, 3(5), 512–519. https://doi.org/10.1158/2159-8290.CD-12-0426
Mondal, G., Lee, J. C., Ravindranathan, A., Villanueva-Meyer, J. E., Tran, Q. T., Allen, S. J., et al. (2020). Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathologica, 139(6), 1071–1088. https://doi.org/10.1007/s00401-020-02155-5
Viaene, A. N., Santi, M., Rosenbaum, J., Li, M. M., Surrey, L. F., & Nasrallah, M. P. (2018). SETD2 mutations in primary central nervous system tumors. Acta Neuropathologica Communications, 6(1), 123. https://doi.org/10.1186/s40478-018-0623-0
Louis, D. N., Perry, A., Wesseling, P., Brat, D. J., Cree, I. A., Figarella-Branger, D., et al. (2021). The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology, 23(8), 1231–1251. https://doi.org/10.1093/neuonc/noab106
Parsons, D. W., Jones, S., Zhang, X., Lin, J. C., Leary, R. J., Angenendt, P., et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science, 321(5897), 1807–1812. https://doi.org/10.1126/science.1164382
Cancer Genome Atlas Research, Brat, D. J., Verhaak, R. G., Aldape, K. D., Yung, W. K., Salama, S. R., et al. (2015). Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. New England Journal of Medicine, 372(26), 2481–98. https://doi.org/10.1056/NEJMoa1402121
Pollack, I. F., Hamilton, R. L., Sobol, R. W., Nikiforova, M. N., Lyons-Weiler, M. A., LaFramboise, W. A., et al. (2011). IDH1 mutations are common in malignant gliomas arising in adolescents: A report from the Children’s Oncology Group. Childs Nervous System, 27(1), 87–94. https://doi.org/10.1007/s00381-010-1264-1
Roux, A., Pallud, J., Saffroy, R., Edjlali-Goujon, M., Debily, M. A., Boddaert, N., et al. (2020). High-grade gliomas in adolescents and young adults highlight histomolecular differences from their adult and pediatric counterparts. Neuro-Oncology, 22(8), 1190–1202. https://doi.org/10.1093/neuonc/noaa024
Turcan, S., Rohle, D., Goenka, A., Walsh, L. A., Fang, F., Yilmaz, E., et al. (2012). IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature, 483(7390), 479–483. https://doi.org/10.1038/nature10866
Johnson, K. C., Anderson, K. J., Courtois, E. T., Gujar, A. D., Barthel, F. P., Varn, F. S., et al. (2021). Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nature Genetics, 53(10), 1456–1468. https://doi.org/10.1038/s41588-021-00926-8
Capper, D., Jones, D. T. W., Sill, M., Hovestadt, V., Schrimpf, D., Sturm, D., et al. (2018). DNA methylation-based classification of central nervous system tumours. Nature, 555(7697), 469–474. https://doi.org/10.1038/nature26000
Castel, D., Philippe, C., Kergrohen, T., Sill, M., Merlevede, J., Barret, E., et al. (2018). Transcriptomic and epigenetic profiling of “diffuse midline gliomas, H3 K27M-mutant” discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathologica Communications, 6(1), 117. https://doi.org/10.1186/s40478-018-0614-1
Hubner, J. M., Muller, T., Papageorgiou, D. N., Mauermann, M., Krijgsveld, J., Russell, R. B., et al. (2019). EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro-Oncology, 21(7), 878–889. https://doi.org/10.1093/neuonc/noz058
Jain, S. U., Rashoff, A. Q., Krabbenhoft, S. D., Hoelper, D., Do, T. J., Gibson, T. J., et al. (2020). H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Molecular Cell, 80(4), 726-735 e7. https://doi.org/10.1016/j.molcel.2020.09.028
Gessi, M., Capper, D., Sahm, F., Huang, K., von Deimling, A., Tippelt, S., et al. (2016). Evidence of H3 K27M mutations in posterior fossa ependymomas. Acta Neuropathologica, 132(4), 635–637. https://doi.org/10.1007/s00401-016-1608-3
Mariet, C., Castel, D., Grill, J., Saffroy, R., Dangouloff-Ros, V., Boddaert, N., et al. (2022). Posterior fossa ependymoma H3 K27-mutant: An integrated radiological and histomolecular tumor analysis. Acta Neuropathologica Communications, 10(1), 137. https://doi.org/10.1186/s40478-022-01442-4
Ryall, S., Guzman, M., Elbabaa, S. K., Luu, B., Mack, S. C., Zapotocky, M., et al. (2017). H3 K27M mutations are extremely rare in posterior fossa group A ependymoma. Childs Nervous System, 33(7), 1047–1051. https://doi.org/10.1007/s00381-017-3481-3
Sievers, P., Sill, M., Schrimpf, D., Stichel, D., Reuss, D. E., Sturm, D., et al. (2021). A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro-Oncology, 23(1), 34–43. https://doi.org/10.1093/neuonc/noaa251
Castelo-Branco, P., Choufani, S., Mack, S., Gallagher, D., Zhang, C., Lipman, T., et al. (2013). Methylation of the TERT promoter and risk stratification of childhood brain tumours: An integrative genomic and molecular study. The lancet Oncology, 14(6), 534–542. https://doi.org/10.1016/S1470-2045(13)70110-4
Dorris, K., Sobo, M., Onar-Thomas, A., Panditharatna, E., Stevenson, C. B., Gardner, S. L., et al. (2014). Prognostic significance of telomere maintenance mechanisms in pediatric high-grade gliomas. Journal of Neuro-oncology, 117(1), 67–76. https://doi.org/10.1007/s11060-014-1374-9
Jafri, M. A., Ansari, S. A., Alqahtani, M. H., & Shay, J. W. (2016). Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med, 8(1), 69. https://doi.org/10.1186/s13073-016-0324-x
Killela, P.J., Reitman, Z.J., Jiao, Y., Bettegowda, C., Agrawal, N., Diaz, L.A., Jr., et al. (2013). TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proceedings of the National Academy of Sciences, 110(15), 6021-6026https://doi.org/10.1073/pnas.1303607110
Karsy, M., Guan, J., Cohen, A. L., Jensen, R. L., & Colman, H. (2017). New Molecular Considerations for Glioma: IDH, ATRX, BRAF, TERT, H3 K27M. Current Neurology and Neuroscience Reports, 17(2), 19. https://doi.org/10.1007/s11910-017-0722-5
Koelsche, C., Sahm, F., Capper, D., Reuss, D., Sturm, D., Jones, D. T., et al. (2013). Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathologica, 126(6), 907–915. https://doi.org/10.1007/s00401-013-1195-5
Bryan, T. M., Englezou, A., Dalla-Pozza, L., Dunham, M. A., & Reddel, R. R. (1997). Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nature Medicine, 3(11), 1271–1274. https://doi.org/10.1038/nm1197-1271
Taylor, K. R., Mackay, A., Truffaux, N., Butterfield, Y., Morozova, O., Philippe, C., et al. (2014). Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nature Genetics, 46(5), 457–461. https://doi.org/10.1038/ng.2925
Paugh, B. S., Zhu, X., Qu, C., Endersby, R., Diaz, A. K., Zhang, J., et al. (2013). Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Research, 73(20), 6219–6229. https://doi.org/10.1158/0008-5472.CAN-13-1491
Paugh, B. S., Broniscer, A., Qu, C., Miller, C. P., Zhang, J., Tatevossian, R. G., et al. (2011). Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. Journal of Clinical Oncology, 29(30), 3999–4006. https://doi.org/10.1200/JCO.2011.35.5677
Pajovic, S., Siddaway, R., Bridge, T., Sheth, J., Rakopoulos, P., Kim, B., et al. (2020). Epigenetic activation of a RAS/MYC axis in H3.3K27M-driven cancer. Nature Communications, 11(1), 6216. https://doi.org/10.1038/s41467-020-19972-7
Maxwell, H. P. (1945). The incidence of interhemispheric extension of glioblastoma multiforme through the corpus callosum. Journal of Neurosurgery, 3, 54–57.
Sharifi, G., Pajavand, A. M., Nateghinia, S., Meybodi, T. E., & Hasooni, H. (2019). Glioma Migration Through the Corpus Callosum and the Brainstem Detected by Diffusion and Magnetic Resonance Imaging: Initial Findings. Frontiers in Human Neuroscience, 13, 472. https://doi.org/10.3389/fnhum.2019.00472
Claes, A., Idema, A. J., & Wesseling, P. (2007). Diffuse glioma growth: A guerilla war. Acta Neuropathologica, 114(5), 443–458. https://doi.org/10.1007/s00401-007-0293-7
Buczkowicz, P., Bartels, U., Bouffet, E., Becher, O., & Hawkins, C. (2014). Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: Diagnostic and therapeutic implications. Acta Neuropathologica, 128(4), 573–581. https://doi.org/10.1007/s00401-014-1319-6
Broniscer, A., & Gajjar, A. (2004). Supratentorial high-grade astrocytoma and diffuse brainstem glioma: Two challenges for the pediatric oncologist. The Oncologist, 9(2), 197–206. https://doi.org/10.1634/theoncologist.9-2-197
Arunachalam, S., Szlachta, K., Brady, S. W., Ma, X., Ju, B., Shaner, B., et al. (2022). Convergent evolution and multi-wave clonal invasion in H3 K27-altered diffuse midline gliomas treated with a PDGFR inhibitor. Acta Neuropathologica Communications, 10(1), 80. https://doi.org/10.1186/s40478-022-01381-0
Hoffman, L. M., DeWire, M., Ryall, S., Buczkowicz, P., Leach, J., Miles, L., et al. (2016). Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: Implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathologica Communications, 4, 1. https://doi.org/10.1186/s40478-015-0269-0
Nikbakht, H., Panditharatna, E., Mikael, L. G., Li, R., Gayden, T., Osmond, M., et al. (2016). Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nature Communications, 7, 11185. https://doi.org/10.1038/ncomms11185
Vinci, M., Burford, A., Molinari, V., Kessler, K., Popov, S., Clarke, M., et al. (2018). Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nature Medicine, 24(8), 1204–1215. https://doi.org/10.1038/s41591-018-0086-7
Federico, S., Brennan, R., & Dyer, M. A. (2011). Childhood Cancer and Developmental Biology: A Crucial Partnership. Current Topics in Developmental Biology, 94, 1–13.
Filbin, M., & Monje, M. (2019). Developmental origins and emerging therapeutic opportunities for childhood cancer (pp. 367–376). Nature Publishing Group.
Deng, Y., Bartosovic, M., Ma, S., Zhang, D., Kukanja, P., Xiao, Y., et al. (2022). Spatial profiling of chromatin accessibility in mouse and human tissues. Nature, 609(7926), 375–383. https://doi.org/10.1038/s41586-022-05094-1
Lu, T., Ang, C. E., & Zhuang, X. (2022). Spatially resolved epigenomic profiling of single cells in complex tissues. Cell, 185(23), 4448-4464 e17. https://doi.org/10.1016/j.cell.2022.09.035
Ziffra, R. S., Kim, C. N., Ross, J. M., Wilfert, A., Turner, T. N., Haeussler, M., et al. (2021). Single-cell epigenomics reveals mechanisms of human cortical development. Nature, 598(7879), 205–213. https://doi.org/10.1038/s41586-021-03209-8
Jessa, S., Blanchet-Cohen, A., Krug, B., Vladoiu, M., Coutelier, M., Faury, D., et al. (2019). Stalled developmental programs at the root of pediatric brain tumors. Nature Genetics, 51(12), 1702–1713. https://doi.org/10.1038/s41588-019-0531-7
Nagaraja, S., Quezada, M. A., Gillespie, S. M., Arzt, M., Lennon, J. J., Woo, P. J., et al. (2019). Histone Variant and Cell Context Determine H3K27M Reprogramming of the Enhancer Landscape and Oncogenic State. Molecular Cell. https://doi.org/10.1016/j.molcel.2019.08.030
Weng, Q., Wang, J., Wang, J., He, D., Cheng, Z., Zhang, F., et al. (2019). Single-Cell Transcriptomics Uncovers Glial Progenitor Diversity and Cell Fate Determinants during Development and Gliomagenesis. Cell Stem Cell, 24(5), 707-723 e8. https://doi.org/10.1016/j.stem.2019.03.006
Funato, K., Major, T., Lewis, P. W., Allis, C. D., & Tabar, V. (2014). Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science, 346(6216), 1529–1533. https://doi.org/10.1126/science.1253799
Ligon, K. L., Alberta, J. A., Kho, A. T., Weiss, J., Kwaan, M. R., Nutt, C. L., et al. (2004). The Oligodendroglial Lineage Marker OLIG2 Is Universally Expressed in Diffuse Gliomas. Journal of Neuropathology & Experimental Neurology, 63, 499.
Lu, Q.R., Park, J.K., Noll, E., Chan, J.A., Alberta, J., Yuk, D., et al. (2001). Oligodendrocyte lineage genes (OLIG) as molecular markers for human glial brain tumors. Proceedings of the National Academy of Sciences, 98, 10851–10856.
Tate, M. C., Lindquist, R. A., Nguyen, T., Sanai, N., Barkovich, A. J., Huang, E. J., et al. (2015). Postnatal growth of the human pons: A morphometric and immunohistochemical analysis (pp. 449–462). Wiley-Liss Inc.
Lindquist, R. A., Guinto, C. D., Rodas-Rodriguez, J. L., Fuentealba, L. C., Tate, M. C., Rowitch, D. H., et al. (2016). Identification of proliferative progenitors associated with prominent postnatal growth of the pons. Nature Communications, 7, 11628. https://doi.org/10.1038/ncomms11628
Filbin, M. G., Tirosh, I., Hovestadt, V., Shaw, M. L., Escalante, L. E., Mathewson, N. D., et al. (2018). Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science, 360(6386), 331–335. https://doi.org/10.1126/science.aao4750
Jessa, S., Mohammadnia, A., Harutyunyan, A. S., Hulswit, M., Varadharajan, S., Lakkis, H., et al. (2022). K27M in canonical and noncanonical H3 variants occurs in distinct oligodendroglial cell lineages in brain midline gliomas. Nature Genetics, 54(12), 1865–1880. https://doi.org/10.1038/s41588-022-01205-w
Misuraca, K. L., Barton, K. L., Chung, A., Diaz, A. K., Conway, S. J., Corcoran, D. L., et al. (2014). Pax3 expression enhances PDGF-B-induced brainstem gliomagenesis and characterizes a subset of brainstem glioma. Acta Neuropathologica Communications, 2, 134. https://doi.org/10.1186/s40478-014-0134-6
Misuraca, K. L., Hu, G., Barton, K. L., Chung, A., & Becher, O. J. (2016). A Novel Mouse Model of Diffuse Intrinsic Pontine Glioma Initiated in Pax3-Expressing Cells. Neoplasia, 18(1), 60–70. https://doi.org/10.1016/j.neo.2015.12.002
Pathania, M., De Jay, N., Maestro, N., Harutyunyan, A. S., Nitarska, J., Pahlavan, P., et al. (2017). H33(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell, 32(5), 684-700 e9. https://doi.org/10.1016/j.ccell.2017.09.014
Cordero, F. J., Huang, Z., Grenier, C., He, X., Hu, G., McLendon, R. E., et al. (2017). Histone H3.3K27M represses p16 to accelerate gliomagenesis in a murine model of DIPG. Molecular Cancer Research, 15, 1243–1254.
Tomita, Y., Shimazu, Y., Somasundaram, A., Tanaka, Y., Takata, N., Ishi, Y., et al. (2022). A novel mouse model of diffuse midline glioma initiated in neonatal oligodendrocyte progenitor cells highlights cell-of-origin dependent effects of H3K27M. Glia, 70, 1681–1698.
Mo, Y., Duan, S., Zhang, X., Hua, X., Zhou, H., Wei, H. J., et al. (2022). Epigenome Programming by H3.3K27M Mutation Creates a Dependence of Pediatric Glioma on SMARCA4. Cancer Discovery, 12(12), 2906–2929. https://doi.org/10.1158/2159-8290.CD-21-1492
Panditharatna, E., Marques, J. G., Wang, T., Trissal, M. C., Liu, I., Jiang, L., et al. (2022). BAF Complex Maintains Glioma Stem Cells in Pediatric H3K27M Glioma. Cancer Discovery, 12(12), 2880–2905. https://doi.org/10.1158/2159-8290.CD-21-1491
Cordero, F. J., Huang, Z., Grenier, C., He, X., Hu, G., McLendon, R. E., et al. (2017). Histone H3.3K27M Represses p16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Molecular Cancer Research, 15(9), 1243–1254. https://doi.org/10.1158/1541-7786.MCR-16-0389
Fortin, J., Tian, R., Zarrabi, I., Hill, G., Williams, E., Sanchez-Duffhues, G., et al. (2020). Mutant ACVR1 Arrests Glial Cell Differentiation to Drive Tumorigenesis in Pediatric Gliomas. Cancer Cell, 37(3), 308-323 e12. https://doi.org/10.1016/j.ccell.2020.02.002
Chen, K.Y., Bush, K., Klein, R.H., Cervantes, V., Lewis, N., Naqvi, A., et al. (2020). Reciprocal H3.3 gene editing identifies K27M and G34R mechanisms in pediatric glioma including NOTCH signaling. Nature Research. https://doi.org/10.1038/s42003-020-1076-0
He, C., Xu, K., Zhu, X., Dunphy, P. S., Gudenas, B., Lin, W., et al. (2021). Patient-derived models recapitulate heterogeneity of molecular signatures and drug response in pediatric high-grade glioma. Nature Communications, 12(1), 4089. https://doi.org/10.1038/s41467-021-24168-8
Monje, M., Mitra, S.S., Freret, M.E., Raveh, T.B., Kim, J., Masek, M., et al. (2011). Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proceedings of the National Academy of Sciences, 108(11), 4453-4458https://doi.org/10.1073/pnas.1101657108
Brabetz, S., Leary, S. E. S., Gröbner, S. N., Nakamoto, M. W., Seker-Cin, H., Girard, E. J., et al. (2018). A biobank of patient-derived pediatric brain tumor models. Nature Medicine, 24, 1752.
du Chatinier, A., Meel, M. H., Das, A. I., Metselaar, D. S., Waranecki, P., Bugiani, M., et al. (2022). Generation of immunocompetent syngeneic allograft mouse models for pediatric diffuse midline glioma. Neuro-Oncology Advances, 4(1), vdac079. https://doi.org/10.1093/noajnl/vdac079
Grasso, C. S., Tang, Y., Truffaux, N., Berlow, N. E., Liu, L., Debily, M. A., et al. (2015). Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nature Medicine, 21(7), 827. https://doi.org/10.1038/nm0715-827a
Lin, G.L., Wilson, K.M., Ceribelli, M., Stanton, B.Z., Woo, P.J., Kreimer, S., et al. (2019). Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Science Translational Medicine, 11(519). https://doi.org/10.1126/scitranslmed.aaw0064
Monje, M., Cooney, T., Glod, J., Huang, J., Baxter, P., Vinitsky, A., et al. (2022). DIPG-10 A Phase I Trial Of Panobinostat Following Radiation Therapy In Children With Diffuse Intrinsic Pontine Glioma (DIPG) Or H3k27m-Mutated thalamic diffuse midline glioma (DMG): report from the pediatric brain tumor consortium (PBTC-047). Neuro-Oncology, 24, i19.
Anastas, J. N., Zee, B. M., Kalin, J. H., Kim, M., Guo, R., Alexandrescu, S., et al. (2019). Re-programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell, 36(5), 528-544 e10. https://doi.org/10.1016/j.ccell.2019.09.005
Pal, S., Kozono, D., Yang, X., Fendler, W., Fitts, W., Ni, J., et al. (2018). Dual HDAC and PI3K Inhibition Abrogates NFκB- and FOXM1-Mediated DNA Damage Response to Radiosensitize Pediatric High-Grade Gliomas. Cancer Research, 78(14), 4007–4021. https://doi.org/10.1158/0008-5472.CAN-17-3691
Bočkaj, I., Martini, T. E. I., De Camargo Magalhães, E. S., Bakker, P. L., Meeuwsen-De Boer, T. G. J., Armandari, I., et al. (2021). The H3.3K27M oncohistone affects replication stress outcome and provokes genomic instability in pediatric glioma. Plos Genetics, 17, e1009868.
Balakrishnan, I., Danis, E., Pierce, A., Madhavan, K., Wang, D., Dahl, N., et al. (2020). Senescence Induced by BMI1 Inhibition Is a Therapeutic Vulnerability in H3K27M-Mutant DIPG. Cell Rep, 33(3), 108286. https://doi.org/10.1016/j.celrep.2020.108286
Senthil Kumar, S., Sengupta, S., Zhu, X., Mishra, D. K., Phoenix, T., Dyer, L., et al. (2020). Diffuse Intrinsic Pontine Glioma Cells Are Vulnerable to Mitotic Abnormalities Associated with BMI-1 Modulation. Molecular Cancer Research, 18(11), 1711–1723. https://doi.org/10.1158/1541-7786.MCR-20-0099
Dhar, S., Gadd, S., Patel, P., Vaynshteyn, J., Raju, G.P., Hashizume, R., et al. (2022). A tumor suppressor role for EZH2 in diffuse midline glioma pathogenesis.BioMed Central 1–14
Wiese, M., Schill, F., Sturm, D., Pfister, S., Hulleman, E., Johnsen, S. A., et al. (2016). No Significant Cytotoxic Effect of the EZH2 Inhibitor Tazemetostat (EPZ-6438) on Pediatric Glioma Cells with Wildtype Histone 3 or Mutated Histone 3.3 (pp. 113–117). Georg Thieme Verlag.
Wiese, M., Hamdan, F. H., Kubiak, K., Diederichs, C., Gielen, G. H., Nussbaumer, G., et al. (2020). Combined treatment with CBP and BET inhibitors reverses inadvertent activation of detrimental super enhancer programs in DIPG cells. Springer Nature.
Chung, C., Sweha, S. R., Pratt, D., Tamrazi, B., Panwalkar, P., Banda, A., et al. (2020). Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas. Cancer Cell, 38(3), 334-349 e9. https://doi.org/10.1016/j.ccell.2020.07.008
Fons, N. R., Sundaram, R. K., Breuer, G. A., Peng, S., McLean, R. L., Kalathil, A. N., et al. (2019). PPM1D mutations silence NAPRT gene expression and confer NAMPT inhibitor sensitivity in glioma. Nature Communications, 10(1), 3790–3810. https://doi.org/10.1038/s41467-019-11732-6
Shen, H., Yu, M., Tsoli, M., Chang, C., Joshi, S., Liu, J., et al. (2020). Targeting reduced mitochondrial DNA quantity as a therapeutic approach in pediatric high-grade gliomas. Neuro-Oncology, 22(1), 139–151. https://doi.org/10.1093/neuonc/noz140
Chheda, Z. S., Kohanbash, G., Okada, K., Jahan, N., Sidney, J., Pecoraro, M., et al. (2018). Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. Journal of Experimental Medicine, 215(1), 141–157. https://doi.org/10.1084/jem.20171046
Mueller, S., Taitt, J.M., Villanueva-Meyer, J.E., Bonner, E.R., Nejo, T., Lulla, R.R., et al. (2022). Mass cytometry detects H3.3K27M-specific vaccine responses in diffuse midline glioma. Journal of Clinical Investigation, 132(12). https://doi.org/10.1172/JCI162283
Mount, C. W., Majzner, R. G., Sundaresh, S., Arnold, E. P., Kadapakkam, M., Haile, S., et al. (2018). Potent antitumor efficacy of anti-GD2 CAR T cells in H3–K27M(+) diffuse midline gliomas. Nature Medicine, 24(5), 572–579. https://doi.org/10.1038/s41591-018-0006-x
Majzner, R. G., Ramakrishna, S., Yeom, K. W., Patel, S., Chinnasamy, H., Schultz, L. M., et al. (2022). GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature, 603(7903), 934–941. https://doi.org/10.1038/s41586-022-04489-4
Haydar, D., Houke, H., Chiang, J., Yi, Z., Ode, Z., Caldwell, K., et al. (2021). Cell-surface antigen profiling of pediatric brain tumors: B7–H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro-Oncology, 23(6), 999–1011. https://doi.org/10.1093/neuonc/noaa278
Grabovska, Y., Mackay, A., O’Hare, P., Crosier, S., Finetti, M., Schwalbe, E. C., et al. (2020). Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nature Communications, 11(1), 4324. https://doi.org/10.1038/s41467-020-18070-y
Lieberman, N. A. P., DeGolier, K., Kovar, H. M., Davis, A., Hoglund, V., Stevens, J., et al. (2019). Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: Implications for development of immunotherapy. Neuro-Oncology, 21(1), 83–94. https://doi.org/10.1093/neuonc/noy145
Ross, J. L., Chen, Z., Herting, C. J., Grabovska, Y., Szulzewsky, F., Puigdelloses, M., et al. (2021). Platelet-derived growth factor beta is a potent inflammatory driver in paediatric high-grade glioma. Brain, 144(1), 53–69. https://doi.org/10.1093/brain/awaa382
Keane, L., Cheray, M., Saidi, D., Kirby, C., Friess, L., Gonzalez-rodriguez, P., et al. (2021). Inhibition of microglial EZH2 leads to anti-tumoral effects in pediatric diffuse midline gliomas. 1–13
Dolma, S., Selvadurai, H. J., Lan, X., Lee, L., Kushida, M., Voisin, V., et al. (2016). Inhibition of Dopamine Receptor D4 Impedes Autophagic Flux, Proliferation, and Survival of Glioblastoma Stem Cells. Cancer Cell, 29(6), 859–873. https://doi.org/10.1016/j.ccell.2016.05.002
Marisetty, A. L., Lu, L., Veo, B. L., Liu, B., Coarfa, C., Kamal, M. M., et al. (2019). REST-DRD2 mechanism impacts glioblastoma stem cell-mediated tumorigenesis (pp. 775–785). Oxford University Press.
Chi, A. S., Tarapore, R. S., Hall, M. D., Shonka, N., Gardner, S., Umemura, Y., et al. (2019). Pediatric and adult H3 K27M-mutant diffuse midline glioma treated with the selective DRD2 antagonist ONC201. Journal of Neuro-Oncology, 145(1), 97–105. https://doi.org/10.1007/s11060-019-03271-3
Stein, M. N., Bertino, J. R., Kaufman, H. L., Mayer, T., Moss, R., Silk, A., et al. (2017). First-in-Human Clinical Trial of Oral ONC201 in Patients with Refractory Solid Tumors. Clinical Cancer Research, 23(15), 4163–4169. https://doi.org/10.1158/1078-0432.CCR-16-2658
Ishizawa, J., Zarabi, S. F., Davis, R. E., Halgas, O., Nii, T., Jitkova, Y., et al. (2019). Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell, 35(5), 721-737 e9. https://doi.org/10.1016/j.ccell.2019.03.014
Graves, P. R., Aponte-Collazo, L. J., Fennell, E. M. J., Graves, A. C., Hale, A. E., Dicheva, N., et al. (2019). Mitochondrial Protease ClpP is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chemical Biology, 14(5), 1020–1029. https://doi.org/10.1021/acschembio.9b00222
Przystal, J. M., Cianciolo Cosentino, C., Yadavilli, S., Zhang, J., Laternser, S., Bonner, E. R., et al. (2022). Imipridones affect tumor bioenergetics and promote cell lineage differentiation in diffuse midline gliomas. Neuro-Oncology, 24(9), 1438–1451. https://doi.org/10.1093/neuonc/noac041
Behjati, S., Tarpey, P. S., Presneau, N., Scheipl, S., Pillay, N., Van Loo, P., et al. (2013). Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nature Genetics, 45(12), 1479–1482. https://doi.org/10.1038/ng.2814
Papillon-Cavanagh, S., Lu, C., Gayden, T., Mikael, L. G., Bechet, D., Karamboulas, C., et al. (2017). Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nature Genetics, 49(2), 180–185. https://doi.org/10.1038/ng.3757
Snuderl, M., Dolgalev, I., Heguy, A., Walsh, M. F., Benayed, R., Jungbluth, A. A., et al. (2019). Histone H3K36I mutation in a metastatic histiocytic tumor of the skull and response to sarcoma chemotherapy. Cold Spring Harbor Molecular Case Studies, 5(5), a004606. https://doi.org/10.1101/mcs.a004606
Fang, D., Gan, H., Lee, J. H., Han, J., Wang, Z., Riester, S. M., et al. (2016). The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science, 352(6291), 1344–8. https://doi.org/10.1126/science.aae0065
Lu, C., Jain, S. U., Hoelper, D., Bechet, D., Molden, R. C., Ran, L., et al. (2016). Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science, 352(6287), 844–849. https://doi.org/10.1126/science.aac7272
Sloan, E. A., Cooney, T., Oberheim Bush, N. A., Buerki, R., Taylor, J., Clarke, J. L., et al. (2019). Recurrent non-canonical histone H3 mutations in spinal cord diffuse gliomas. Acta Neuropathologica, 138(5), 877–881. https://doi.org/10.1007/s00401-019-02072-2
Boileau, M., Shirinian, M., Gayden, T., Harutyunyan, A. S., Chen, C. C. L., Mikael, L. G., et al. (2019). Mutant H3 histones drive human pre-leukemic hematopoietic stem cell expansion and promote leukemic aggressiveness. Nature Communications, 10(1), 2891. https://doi.org/10.1038/s41467-019-10705-z
Lehnertz, B., Zhang, Y. W., Boivin, I., Mayotte, N., Tomellini, E., Chagraoui, J., et al. (2017). H3(K27M/I) mutations promote context-dependent transformation in acute myeloid leukemia with RUNX1 alterations. Blood, 130(20), 2204–2214. https://doi.org/10.1182/blood-2017-03-774653
Bennett, R. L., Bele, A., Small, E. C., Will, C. M., Nabet, B., Oyer, J. A., et al. (2019). A Mutation in Histone H2B Represents a New Class of Oncogenic Driver. Cancer Discovery, 9(10), 1438–1451. https://doi.org/10.1158/2159-8290.CD-19-0393
Nacev, B. A., Feng, L., Bagert, J. D., Lemiesz, A. E., Gao, J., Soshnev, A. A., et al. (2019). The expanding landscape of “oncohistone” mutations in human cancers. Nature, 567(7749), 473–478. https://doi.org/10.1038/s41586-019-1038-1
Bagert, J. D., Mitchener, M. M., Patriotis, A. L., Dul, B. E., Wojcik, F., Nacev, B. A., et al. (2021). Oncohistone mutations enhance chromatin remodeling and alter cell fates. Nature Chemical Biology, 17(4), 403–411. https://doi.org/10.1038/s41589-021-00738-1
Feinberg, A. P., & Vogelstein, B. (1983). Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature, 301(5895), 89–92. https://doi.org/10.1038/301089a0
Hanahan, D. (2022). Hallmarks of Cancer: New Dimensions. Cancer Discovery, 12(1), 31–46. https://doi.org/10.1158/2159-8290.CD-21-1059
Acknowledgements
The authors acknowledge support from the National Cancer Institute (CA096832 to SJB, CA265285 to KB, and CA271570 to JTR) and the American Lebanese Associated Charities. We thank members of the Baker lab and Dr. David Ellison for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ocasio, J.K., Budd, K.M., Roach, J.T. et al. Oncohistones and disrupted development in pediatric-type diffuse high-grade glioma. Cancer Metastasis Rev 42, 367–388 (2023). https://doi.org/10.1007/s10555-023-10105-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-023-10105-2