
Abstract
Recent genomic studies of pediatric high-grade gliomas (pHGGs) have unraveled important new clues into the pathogenesis of these tumors. One novel insight that was previously unappreciated is the role of epigenetic alterations in pediatric gliomagenesis. This was realized when mutations in histone 3.3/3.1 (H3.3/H3.1) were identified in these tumors. A related concept is that certain genetic alterations only occur in a region-specific manner within the central nervous system. As an example, K27M H3.3/H3.1 mutations are present in high-grade gliomas that arise along the midline of the central nervous system while G34R and G34V H3.3 mutations are present in high-grade gliomas that arise in the cerebral cortex. In addition, the realization that gliomas that harbor histone mutations are mutually exclusive of isocitrate dehydrogenase (IDH) mutant gliomas, which primarily arise in adults, has reinforced the notion that pediatric gliomas and adult gliomas are biologically disparate diseases. Incorporating this new data within the framework of our existing knowledge of the genetic alterations of pHGGs gives new hope that we will successfully identify effective therapies for these tumors in the upcoming decade.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
The central nervous system (CNS) is the second most common location for tumorigenesis in children with the majority of tumors being benign. Pediatric high-grade gliomas (pHGGs) only account for approximately 8–12 % of all childhood CNS tumors but are a leading cause of mortality in children [1, 2]. pHGG histologies include anaplastic astrocytomas (World Health Organization or WHO grade III), anaplastic oligodendrogliomas (WHO grade III), and glioblastomas/gliosarcomas (WHO grade IV). pHGGs can arise anywhere in the CNS and those that arise in the brainstem are also called diffuse intrinsic pontine glioma or DIPG. Unfortunately, little therapeutic progress has been made over the last few decades, and clinical outcomes for these patients remain dismal. To date the only effective modality is focal radiation, which provides only temporary tumor control in most patients. One of the reasons for this lack of clinical progress has been our limited understanding of the molecular genetics and tumor biology of pHGGs. Very recently, next generation sequencing studies have led to the discovery of a fundamentally novel class of genetic alterations, implicating epigenetic reprogramming as a central component of pHGG pathogenesis and opening potential avenues for new, biology-based treatment approaches.
Additional key insights from recent genomic studies of pHGG include the appreciation of molecular genetic heterogeneity not only between patients, but also within the tumor tissue of the same patients, distinct biological differences compared to adult high-grade gliomas, and the recognition of differences in tumorigenesis based on age and tumor location within the CNS. Clearly, these insights will need to be taken into account in developing novel therapeutic strategies that will hopefully lead to improved outcomes of patients afflicted with these highly aggressive and lethal tumors. In this chapter, we will briefly describe the histopathology, cytogenetic, molecular genetic and epigenetic alterations, as well as gene expression profiling of pHGGs. We will also discuss key signaling pathways pertinent to the advancement of molecular targeted therapies in pHGGs. Lastly, we will review current clinical trials of molecular targeted therapies for pHGGs and emerging future avenues of clinical investigation.
Histopathology
High-grade gliomas consist of the gliomas graded by the World Health Organization as Grades III and IV. As such, the following tumors fall into this group: anaplastic astrocytoma, glioblastoma multiforme (GBM), anaplastic oligodendroglioma, and anaplastic oligo-astrocytoma. While not a strictly histogenetic type, anaplastic glioma has been assigned to those cases that do not clearly fall into either anaplastic astrocytoma or anaplastic oligodendroglioma due to small sample size, therapeutic effect, or some other histologic artifact that renders specific classification difficult.
High-grade gliomas of all types share common features of high mitotic activity, vascular endothelial proliferation and, quite commonly, focal necrosis. Oligodendrogliomas share features with the myelinating cells of the CNS, the oligodendrocytes. These include round, regular nuclei and clear cytoplasm that ring the nuclei producing a fried-egg appearance. Anaplastic oligodendrogliomas exhibit dense hypercellularity, frequent mitotic figures, vascular endothelial proliferation, and foci of necrosis. These foci may or may not be ringed by cells tightly surrounding the focus, defined as pseudopalisading necrosis (Fig. 7.1).

Pediatric glioblastoma exhibiting pseudopalisading necrosis. Hematoxylin and eosin staining of a Grade IV GBM (40× magnification). Arrows point to pseudopalisading necrosis
Anaplastic astrocytomas differ from anaplastic oligodendrogliomas by having hyperchromatic nuclei that are often elongated, irregular, or smudged. Mitotic figures may not be common, but can be found by immunohistochemical markers of proliferation such as the MIB-1 index. While the tumor cells are not as densely packed as in the oligodendroglioma, they are nonetheless, obviously more hypercellular than the white matter by a factor of three- to tenfold. Vascular changes are frequent in anaplastic astrocytoma, but the defining element of vascular proliferation as found in the Grade IV glioblastoma, is debated. While most brain tumor neuropathologists agree that endothelial duplication circumferentially ringing the vessel is a feature of glioblastomas, it is unclear where glomeruloid vessels and vascular garlands or loops of vessels fall in astrocytoma grading. Among astrocytic tumors, foci of necrosis are reserved for glioblastomas.
Glioblastoma multiforme, the most malignant and most common of the high-grade gliomas, is an astrocytic neoplasm that has been found to most commonly arise de novo without a preceding history of glioma, with only 5 % of glioblastomas occurring as a result of progression from a lower grade astrocytoma. Both de novo and progressive GBMs exhibit nuclear pleomorphism, mitotic activity, vascular proliferation, and/or necrosis. While densely cellular in some foci of the tumor, there is commonly an infiltrative component that can be found distant from the central mass. This infiltrative capability currently makes GBMs impossible to cure by surgery or local–regional radiation therapy.
Epigenetics
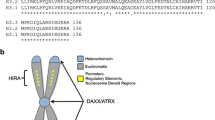
With the advent of next generation sequencing, our understanding of the genetic alterations in pHGGs has been turned upside down. This technological advance has led to the discovery of heterozygous K27M or G34R/V mutations in the tail of histone 3.3 or K27M mutations in the tail of histone 3.1 and loss of function mutations in the chromatin remodelers ATRX (α-thalassaemia/mental retardation syndrome X-linked) and DAXX (death-domain associated protein) in pHGGs (Fig. 7.2; [3, 4]). As a brief review, the fundamental repeating unit of chromatin is the nucleosome, which consists of approximately 147 bp of superhelical DNA wrapped around the radial surface of an octamer of highly conserved core histone proteins (two copies of each H2A, H2B, H3, and H4). Histone proteins are subject to a wide array of covalent modifications that occur primarily at amino (N−) and carboxy (C−) termini. The tail regions of core histones contain flexible and highly basic amino acid sequences that are highly conserved and serve as substrates for several posttranslational modifications such as acetylation, methylation, ADP-ribosylation, ubiquitylation, and phosphorylation. These modifications impact gene transcription, DNA replication, and chromatin assembly. The histone code states that distinct patterns of histone modifications act in concert with DNA methylation, noncoding RNAs, and transcription factors to generate “histone-epigenetic codes” that are read by effector proteins [5]. Lastly, another level of complexity is histone variants (e.g., H3.3 vs. H3.1), which are relevant to pHGGs. Although the difference between H3.3 and H3.1 is only four amino acids, H3.1 (also called canonical core H3) is only incorporated into nucleosomes during the S-phase of the cell cycle while H3.3 incorporation into nucleosomes is cell-cycle independent. Furthermore, ATRX and DAXX are both H3.3 chaperones and together they facilitate the deposition and remodeling of H3.3 containing nucleosomes [6].

Epigenetic alterations associated with pediatric high-grade gliomagenesis. This schematic of the H3.3/H3.1 tail illustrates the three types of epigenetic mutations seen in pHGG: (1) K27M mutations which impact H3K27 methylation, (2) G34R/V or SETD2 mutations, both of which impact H3K36 methylation, and (3) ATRX/DAXX mutations which likely impact H3.3 deposition and alternative lengthening of telomeres
The initial two manuscripts describing these mutations noted, in the brainstem there are K27M mutations in either H3.3 or H3.1 in up to 80 % of DIPGs, while the G34R/V mutations are found only in H3.3 and were primarily found in pHGGs located in the cerebral cortex. In addition, G34R/V mutations co-occur with loss of function ATRX or DAXX mutations and are associated with the ALT (alternative lengthening of telomeres) phenotype [3, 4, 7, 8]. In a follow-up paper, Sturm and colleagues [9] pursue an integrative approach based on epigenetic, copy number, expression, and genetic analyses on over 200 adult and pediatric GBMs to identify six distinct DNA methylation clusters which were labeled as “IDH,” “K27,” “G34,” “receptor tyrosine kinase (RTK) I (platelet-derived growth factor receptor A or PDGFR-A),” “mesenchymal,” and “RTK II (Classic).” A key finding of this analysis was that H3F3A K27 and H3F3A G34 mutations were exclusively distributed to the K27 and G34 clusters, respectively, and these were mutually exclusive of the isocitrate dehydrogenase (IDH1) mutations. The RTK I (PDGFR-A) group had a high frequency of PDGFR-A amplification and the RTK II Classic group had a very high frequency of whole chromosome seven gain, whole chromosome ten loss, frequent deletion of cyclin-dependent kinase inhibitor 2a (CDKN2A), and amplification of epidermal growth factor receptor (EGFR). This RTK II Classic subgroup is completely devoid of pediatric patients. Remarkably, the clusters are associated with patient age, with K27M patients being the youngest (median age 10.5; range 5–23) and G34 patients being the second youngest (median age 18 years; range 9–42). The RTK I “PDGFR-A” cluster (median age 36 years, range 8–74 years) and the IDH cluster (median age 40 years, range 13–71 years) mostly comprised young adults, while the oldest cluster, the RTK II “Classic” cluster, comprised older adults (median age 58, range 36–81 years). The other remarkable finding was that the epigenetic GBM subgroups showed region-specific predilection within the CNS whereby the K27-mutant tumors were predominantly seen in midline locations such as the thalamus, pons, and the spinal cord while the tumors in the other five subgroups almost exclusively arose in the cerebral hemispheres. This remarkable observation clearly suggests that the mechanism of gliomagenesis is distinct in different regions of the CNS. Lastly, these subgroups also correlated with survival with the K27 subgroup having the shortest survival, the IDH subgroup with the longest survival, and the other subgroups in between. Interestingly, both the IDH and H3F3A mutations co-occur with p53 mutations, suggesting that p53 mutations do not have independent prognostic significance [9].
How histone mutations contribute to pHGG pathogenesis is subject to current investigations, but Lewis and colleagues recently reported an initial glimpse into the mechanism. They reported that the K27M H3.3/H3.1 mutations inhibit polycomb repressive complex 2 (PRC2), the enzyme complex that adds methyl groups to H3 lysine 27. Under normal circumstances, this histone mark is repressive, and inhibition of PRC2 results in global loss of H3 lysine 27 trimethylation. The mechanism of the G34R/V histone mutations is less well understood, but results in a local decrease in H3 lysine 36 trimethylation [10]. In addition to the epigenetic mutations described above, pHGGs recently have also been reported to harbor loss of function mutations in SETD2, an H3K36 trimethyltransferase. Perhaps not surprisingly, SET2D mutations were mutually exclusive with H3F3A mutations, but they did overlap with IDH1 mutations [11]. High-grade gliomas with SET2D mutations were found exclusively in tumors arising in the cerebral hemispheres, reinforcing the notion that H3K36 is important for gliomagenesis in the cerebral hemispheres, while H3K27 is central in the etiology of tumors arising in midline structures of the CNS. Lastly, while IDH mutations are primarily found in adult gliomas, adolescents 13 years old and older can also harbor activating mutations in IDH1 in amino acid 132 [12, 13]. These IDH1 mutations have also been reported to impact histone marks, but through a completely different mechanism [14].
Cytogenetics
Several studies have investigated the spectrum of copy number aberrations in pHGGs [15–22]. Copy number changes in pHGGs are best subdivided between broad chromosomal gains and losses and focal gains and losses. Broad low-amplitude gains of chromosome 1q were identified as well as broad losses of 10q, 13q, and 14q. Focal gains have been reported in PDGFR-A, cyclin D1-3 (CCND1-3), cyclin-dependent kinase 4 (CDK4), cyclin-dependent kinase 6 (CDK6), MYC, v-myc avian myelocytomatosis viral oncogene neuroblastoma-derived homolog (MYCN), EGFR, V-Erb-A Avian Erythroblastic Leukemia Viral Oncogene Homolog-Like 4 (ERBB4), MET, hepatocyte growth factor (HGF), insulin-like growth factor-1 receptor (IGF1R), insulin-like growth factor 2 (IGF2), platelet-derived growth factor B (PDGFB), Neuregulin 1 (NRG1), phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA), PIK3C2B, PIK3C2G, PIK3R5, Kirsten rat sarcoma viral oncogene homolog (KRAS), v-akt murine thymoma viral oncogene homolog 1 (AKT1), AKT3, S6K1, and murine double minute 4 (MDM4). The most common homozygous focal loss was at CDKN2A/CDKN2B. Other homozygous focal losses included the following genes: CDKN2C, neurofibromin-1 (NF1), PTEN (phosphatase and tensin homolog), retinoblastoma (RB1), TP53, TP73, and protein tyrosine phosphatase receptor type D (PTPRD). Interestingly, copy number alterations are also age- and region specific. PDGFR-A is the most common amplified receptor tyrosine kinase (RTK) in pHGGs while EGFR amplification is the most common amplified RTK in adult high-grade glioma. Gains of chromosomes 2q, 8q, and 9q and losses of 16q, 17p, and 20p were significantly more frequent in DIPG than in non-brainstem pHGGs. Furthermore, focal deletions of CDKN2A are extremely rare in DIPGs and are found in 26 % of non-brainstem pHGGs [23]. Lastly, Barrow et al. described homozygous loss of ADAM3A in 16 % of pHGGs, although the function of this gene and how its loss contributes to pediatric gliomagenesis is not known [17]. Figure 7.3 includes a summary figure of the genetic alterations in pHGGs, as well as a table, which lists the genetic alterations in DIPG, non-brainstem pHGG, and adult HGG.

Genetic alterations implicated in pHGGs. Genetic alterations vary by location and age. (a) Genetic alterations observed in pHGGs located in the cerebral cortex, or in midline areas of the CNS, or common to all pHGGs. Arrows pointing to midline areas from top to bottom: thalamus, pons, and spinal cord. (b) Distinct molecular genetics of DIPG as compared to pHGGs (non-DIPG) and adult high-grade gliomas. Table is adapted from: Kristin M Schroeder, Christine M Hoeman, and Oren J Becher, Children are not just little adults: recent advances in understanding of diffuse intrinsic pontine glioma biology, Pediatric Research, 2014 [57]
Gene Expression
Gene expression profiling, a method to analyze the mRNA expression of all genes in the tumor, is another useful technique to study the complex biology of cancer. In fact, mRNA analysis of a select number of genes is used to make clinical decisions in some types of breast cancer. In pHGGs, unsupervised hierarchical clustering identified three main tumor subgroups: HC1/proliferative, HC2/proneural, and HC3/mesenchymal [15]. Gene ontology analysis across the groups revealed that HC1 is most associated with cell-cycle genes; HC2 is most associated with neuronal differentiation, while HC3 is most associated with extracellular matrix–receptor interactions and cell adhesion. Interestingly, HC1 is most associated with amplifications targeting the PDGFR signaling cascade, which ties together PDGF signaling with cell growth. If one were to compare the expression profiling of pHGGs to adult high-grade gliomas, PDGFR-A mRNA is significantly overexpressed in pHGGs relative to adult high-grade gliomas while EGFR mRNA is significantly repressed in pHGGs relative to adult high-grade gliomas. With regard to the HC3/mesenchymal group, immune response genes were also enriched and more specifically associated with microglia/macrophages and monocytes [24]. Lastly, using principal component analysis (PCA), two independent groups noted that the expression profiling of DIPGs consists of a distinct cluster separate from non-brainstem gliomas, reinforcing the notion of region-specific differences in pediatric CNS gliomagenesis [23, 25].
Prognostic Stratification
Until recently, the concept that HGGs comprise several, biologically distinct diseases associated with age and location was not fully appreciated. As a consequence, current research efforts center on developing a better understanding of tumor biology and accordingly, devise appropriate classification schemes. Similar to the increased stratification of leukemias in children, molecular stratification of pHGG will continue to become increasingly refined, in parallel with advances in our understanding of disease biology. The ongoing challenge is how to best incorporate new molecular prognostic factors with well-established prognostic factors, such as extent of resection and tumor grade [2]. For over a decade, p53 overexpression has been recognized as a poor prognostic factor in pHGGs. Most importantly, this association was found to be independent of age, histologic features, the extent of resection, or tumor location [26]. Overexpression of p53 as determined by immunohistochemistry, however, is an imperfect proxy for oncogenic p53 mutations, and taken in context with our current knowledge of the molecular genetics of HGG, a clearer picture emerges. For example, as previously mentioned in the epigenetics section of this chapter, IDH mutations frequently co-occur with p53 mutations, and these tumors currently have the best prognosis. However, p53 mutations also overlap with K27M mutations, which appear to have the worst prognosis. According to retrospective studies, K27M is a poor prognostic factor in pediatric GBM, although it is unclear whether this is due to a different biology versus the midline location of these tumors, which limits surgical options [9, 27]. Within the K27M subgroup, DIPGs have the worst prognosis, with a median survival time of 9–12 months and greater than 90 % of children dying within 2 years [28]. Lastly, PTEN mutations or loss of PTEN expression by IHC have been reported to be significantly associated with decreased survival in pHGGs, but this has so far only been reported in small cohorts and will require further validation [29, 30].
Molecular Signaling Pathways
Three pathways that are most implicated in pHGG pathogenesis are the p53, retinoblastoma protein (Rb), and RTK/Ras/phosphoinositide 3-kinase (PI3K) signaling pathways. These pathways are dysregulated in both pediatric and adult high-grade gliomas, and the importance of these three pathways in adult gliomagenesis was recently underscored by the mutual exclusivity of alterations affecting these pathways [31]. Evolving knowledge of precisely how these pathways contribute to tumor initiation and growth is expected to lead to better-informed molecular targeted therapeutic approaches. With regard to activation of the RTK/Ras/PI3K pathway, 80 % of pHGGs activate this pathway through amplification of RTKs, and/or activating mutations in PI3K, and/or loss of PTEN either through deletion or promoter methylation [32]. Below is a brief summary of the key molecular signaling pathways.
RTK/Ras/PI3K Pathway
The axis of PI3K signaling in cancer begins with engagement of growth factors by RTKs such as PDGFR, MET, EGFR, and IGF-1R (Fig. 7.4). PI3K, a lipid kinase, is then recruited to plasma membrane-anchored receptors, is activated, and phosphorylates PIP2 to generate PIP3. Through its pleckstrin homology (PH) domain, the nodal kinase AKT (also known as PKB) binds to PIP3, where it is activated by two phosphorylation events, and triggers a complex cascade of signals that regulate growth, proliferation, survival, and motility. The lipid phosphatase, PTEN, antagonizes this process by dephosphorylating PIP3 to inhibit activation of AKT. PI3K is activated downstream of numerous RTKs that directly, or through adaptor proteins, bind and activate PI3K. PI3K activity is thus carefully regulated by growth factor–receptor interactions. In fact, the vast majority of PI3K remains inactive in the cytoplasm, removed from its plasma membrane-associated substrates, and only a small percentage of PI3K becomes activated upon growth factor stimulation. Therefore, even slight modulations in receptor activity can lead to many-fold increases in PI3K activity [33].

RTK/Ras/PI3K pathway. Growth factors such as EGF, PDGF, HGF, and IGF2 engage with their respective RTKs leading to PI3K activation. PI3K activation phosphorylates PIP2 to generate PIP3. AKT binds to PIP3, becomes activated, and triggers a complex cascade of signals that regulate growth, proliferation, survival, and motility. PTEN, a naturally occurring antagonist of this pathway, dephosphorylates PIP3 inhibiting activation of AKT. Growth factors and RTK interactions also regulate cell proliferation and survival through activation of Ras followed by sequential activation of Raf, Mek, and Erk. Starred (asterisk) factors mutated in RTK/Ras/PI3K pathway are prevalent in pHGGs
In addition to copy number alterations, pHGGs can also harbor additional alterations in the RTK/Ras/PI3K pathways through somatic mutations or alternative splicing. The most commonly mutated RTK in pHGGs is PDGFR-A where mutations in the extracellular domain as well as in the tyrosine kinase domain have been described in approximately 10 % of these tumors [3, 25, 34]. By contrast, the most commonly mutated RTK in adult high-grade glioma is EGFR. EGFRvIII (an EGFR lacking exons 2–7 resulting in ligand-independent signaling) is an alternatively spliced EGFR variant found in 19 % of adult HGGs, but has also been reported in 17 % of pHGGs [31, 35]. Downstream of RTKs, activating mutations in PIK3CA have been described in a subset of pHGGs both inside and outside of the brainstem [36, 37]. While activating Ras mutations have rarely been described in pHGGs (G12V Kras reported by Schiffman et al. [16]), loss of function NF1 (neurofibromatosis type 1 which negatively regulates the Ras pathway) mutations has been identified in a subset of pHGGs outside of the brainstem [3]. Furthermore, activating V600E Braf mutations have also been described in pHGGs [11, 16, 38, 39]. V600E Braf mutations can also be found in low-grade gliomas, but usually in isolation, suggesting that cooperating mutations (such as CDKN2A) may be required for a high-grade phenotype in V600E Braf mutant tumors [16].
RB Pathway
The retinoblastoma protein (RB) is a tumor suppressor protein and key regulator of cell-cycle control. The RB pathway consists of five families of proteins: CDKN (e.g., Ink4a), D-type cyclins, D-cyclin-dependent protein kinases (cdk4, cdk6), RB-family of pocket proteins (RB, p107, p130), and the E2F-family of transcription factors. Each Ink4-protein (p16Ink4a, p15Ink4b, and p18Ink4c) can bind to and inhibit the activity of cdk4 and cdk6. Each D-cyclin protein can associate with cdk4 or cdk6 to form the active kinase complex. Therefore, Ink4 proteins compete with the D-cyclins for cdk4/6 to prevent the formation of the active kinase complex [40]. Importantly, proteins in this pathway are commonly altered in pHGGs, primarily through copy number aberrations: cdkn2a deletions and/or amplification of cdk4, cdk6, cyclin d1, d2, and d3 [15, 19, 23].
The RB pathway regulates cell proliferation as the proteins in this pathway are activated and/or inhibited by growth-promoting, as well as growth-suppressing signals (Fig. 7.5). During regulated cell proliferation, as cells respond to mitogenic signals and commit to cell-cycle entry the complex of D-cyclin/cdk4/6 is activated. The primary cellular targets of the D-cyclin/cdk4/6 complex are the RB-family of pocket proteins (henceforward referred to as RB), which inhibit transcription by directly inhibiting the activity of E2F. Hyperphosphorylation of RB by activated D-cyclin/cdk4/6 complexes renders RB inactive and in turn allows for the release of E2F transcription factors, leading to the activation of E2F target genes involved in cell-cycle progression [40]. Recently, a preclinical trial using genetically engineered mouse models identified a population of pHGG patients that may be sensitive to treatment with a highly selective cdk4/6 inhibitor [41]. Inhibition of cdk4/6 in murine high-grade gliomas harboring CDKN2A loss provided a significant survival benefit and holds promise for translation into clinical trials.

RB pathway. The RB pathway is an important regulator of cell-cycle control. Cyclin D1 and cdk4/6 bind to form a complex that phosphorylates RB. In its active form RB is bound to E2F transcription factors. Hyperphosphorylation of RB renders it inactive and allows for the release of E2F transcription factors. E2F transcription factors activate E2F target genes, leading to initiation of S-phase and cell-cycle progression. CDKN (e.g., INK4a) is a tumor suppressor gene that inactivates the cyclin D1/cdk4/6 complex. Cdkn2a deletions and/or amplification of cdk4, cdk6, cyclin D1, D2, or D3 are common in pHGGs
P53 Pathway
The p53 protein plays a key role in eliciting cellular responses to a variety of stress signals, such as DNA damage, hypoxia, and aberrant proliferative signals such as oncogene activation. Following cellular stresses, p53 is stabilized and binds to DNA as a tetramer, in a sequence-specific manner that results in the transcriptional regulation of genes involved in DNA repair, cell-cycle arrest, senescence, and apoptosis [42]. The critical role of this gene in tumor suppression in pHGG is clear as evidenced by the abundant, inactivating somatic mutations, which were recently reported in as many as 77 % of DIPGs [27]. Besides p53 mutations, other mechanisms to inactivate the p53 pathway include amplification of mouse double minute 2 homolog (MDM2) and MDM4. MDM2 is an important negative regulator of p53 working through two mechanisms: It is an E3 ubiquitin ligase that targets p53 for proteosomal degradation and it can also inhibit p53 transcriptional activation. MDM4 is a homolog of MDM2 and can also inhibit p53 transcriptional activity. In pHGGs, MDM2 is overexpressed but it is not amplified while MDM4 amplifications have been observed [15, 19, 43]. Interestingly, p53 mutations were reported to occur significantly more often in pediatric GBM relative to adult GBM [3]. This may be related to the fact that CDKN2A deletions are significantly more common in adult GBMs. CDKN2A encodes two transcripts: Ink4a (an endogenous cdk4/6 inhibitor discussed in the RB section) and alternative reading frame (ARF). ARF inhibits p53 degradation by sequestering MDM2 to the nucleolus and rendering it inactive. In summary, the p53 pathway is inactivated in the majority of pHGGs primarily through p53 mutations but also through MDM2 overexpression, MDM4 amplification, and CDKN2A loss, preventing the tumor cells from responding appropriately to cellular stresses.
Molecular Targeted Therapies
Despite recent advancements in our knowledge of key molecular alterations in pHGGs, this new depth of understanding has not translated into improved clinical therapies thus far. There have been numerous clinical trials with molecular targeted therapies for children with high-grade gliomas, but none have been demonstrated to significantly prolong survival. Most of the targeted therapy trials to date have focused on targeting RTKs (the upstream part of the RTK/Ras/PI3K pathway), and angiogenesis (VEGF or αv integrins). The lack of efficacy is likely due to activation of feedback loops, redundant activation of RTK pathways in glioma [44], intratumoral heterogeneity [15], and potentially inadequate drug delivery across the blood–brain barrier [45]. The following is not an exhaustive list of all targeted therapies that have been evaluated in pHGGs, but a brief description of some of the most relevant studies.
There have been multiple studies evaluating EGFR inhibitors (erlotinib, lapatinib, gefitinib) in pHGGs and none of them have demonstrated significant efficacy, even though the target has been demonstrated as present in a subset of pHGGs ([13, 45–49]. Other studies evaluating molecular targeted therapies in pHGGs include evaluation of inhibitors of PDGFR (imatinib), mTOR (temsirolimus), Ras (tipifarnib), VEGF (bevacizumab), αv integrin antagonist (cilengitide), Notch (MK-0752), and VEGFR2 (vandetanib) without success [47, 50–55]. Recently, a combination study of vandetanib and dasatinib (PDGFR inhibitor) was also reported with limited success [56]. Interestingly, the authors noted a 2 % cerebrospinal fluid to plasma exposure in two of the patients in the study, suggesting that inadequate drug delivery may explain the lack of response. Adequate drug delivery across the blood–brain barrier remains an obstacle in pHGG, and particularly in DIPG.
Future Directions
It is our hope that advances in our understanding of the genetic alterations of pHGGs will eventually translate into improved therapies. There is a great deal of excitement surrounding the discovery of highly specific histone mutations in pHGGs, and it remains to be seen how one can target such genetic alterations therapeutically. So far, there are two classes of epigenetic drugs that have been FDA approved for cancer: histone deacetylase (HDAC) inhibitors for cutaneous T-cell lymphoma and DNA methyltransferase (DNMT) inhibitors for myelodysplastic syndrome. In addition, there are numerous new classes of epigenetic drugs that have shown promise in preclinical trials and have recently entered clinical trials such as bromodomain inhibitors (a bromodomain is a protein domain that can bind an acetylated lysine) and enhancer of zeste 2 (EZH2) inhibitors. Furthermore, there are new therapeutic targets that are currently being evaluated in clinical trials for children with high-grade gliomas such as inhibitors of the enzyme poly (ADP-ribose) polymerase (PARP), inhibitors of telomerase (Imetelstat), and V600E Braf inhibitors. In summary, there are numerous new promising therapeutic targets in pHGGs, and the challenge is how to prioritize the translation of novel agents into clinical trials in children with high-grade gliomas and how to combine these novel agents synergistically. Undoubtedly, deeper insights into the biology of pHGGs will continue to emerge over the next years, opening new therapeutic avenues. The inter-patient heterogeneity of the genetic alterations in pHGGs implies that more personalized approaches may be needed, and a current V600E Braf inhibitor pediatric study with dabrafenib (ClinicalTrials.gov NCT01677741) is one example in the right direction, as only patients whose tumors harbor a V600E Braf mutation are allowed to enroll.
References
Fangusaro J. Pediatric high grade glioma: a review and update on tumor clinical characteristics and biology. Front Oncol. 2012;2:105.
Finlay JL, et al. Randomized phase III trial in childhood high-grade astrocytoma comparing vincristine, lomustine, and prednisone with the eight-drugs-in-1-day regimen. Childrens Cancer Group. J Clin Oncol. 1995;13(1):112–23.
Schwartzentruber J, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–31.
Wu G, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44(3):251–3.
Maze I, Noh KM, Allis CD. Histone regulation in the CNS: basic principles of epigenetic plasticity. Neuropsychopharmacology. 2013;38(1):3–22.
Lewis PW, et al. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A. 2010;107(32):14075–80.
Heaphy CM, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333(6041):425.
Nguyen DN, et al. Molecular and morphologic correlates of the alternative lengthening of telomeres phenotype in high-grade astrocytomas. Brain Pathol. 2013;23(3):237–43.
Sturm D, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–37.
Lewis PW, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340(6134):857–61.
Fontebasso AM, et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013;125(5):659–69.
Yan H, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73.
Pollack IF, et al. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children’s Oncology Group. Childs Nerv Syst. 2011;27(1):87–94.
Losman JA, Kaelin Jr WG. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27(8):836–52.
Paugh BS, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010;28(18):3061–8.
Schiffman JD, et al. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res. 2010;70(2):512–9.
Barrow J, et al. Homozygous loss of ADAM3A revealed by genome-wide analysis of pediatric high-grade glioma and diffuse intrinsic pontine gliomas. Neuro Oncol. 2011;13(2):212–22.
Bax DA, et al. A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin Cancer Res. 2010;16(13):3368–77.
Warren KE, et al. Genomic aberrations in pediatric diffuse intrinsic pontine gliomas. Neuro Oncol. 2012;14(3):326–32.
Zarghooni M, et al. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol. 2010;28(8):1337–44.
Wong KK, et al. Genome-wide allelic imbalance analysis of pediatric gliomas by single nucleotide polymorphic allele array. Cancer Res. 2006;66(23):11172–8.
Phillips JJ, et al. PDGFRA amplification is common in pediatric and adult high-grade astrocytomas and identifies a poor prognostic group in IDH1 mutant glioblastoma. Brain Pathol. 2013;23(5):565–73.
Paugh BS, et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol. 2011;29(30):3999–4006.
Engler JR, et al. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS One. 2012;7(8):e43339.
Puget S, et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One. 2012;7(2):e30313.
Pollack IF, et al. Expression of p53 and prognosis in children with malignant gliomas. N Engl J Med. 2002;346(6):420–7.
Khuong-Quang DA, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124(3):439–47.
Freeman CR, Perilongo G. Chemotherapy for brain stem gliomas. Childs Nerv Syst. 1999;15(10):545–53.
Raffel C, et al. Analysis of oncogene and tumor suppressor gene alterations in pediatric malignant astrocytomas reveals reduced survival for patients with PTEN mutations. Clin Cancer Res. 1999;5(12):4085–90.
Thorarinsdottir HK, et al. Protein expression of platelet-derived growth factor receptor correlates with malignant histology and PTEN with survival in childhood gliomas. Clin Cancer Res. 2008;14(11):3386–94.
Brennan CW, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–77.
Mueller S, et al. PTEN promoter methylation and activation of the PI3K/Akt/mTOR pathway in pediatric gliomas and influence on clinical outcome. Neuro Oncol. 2012;14(9):1146–52.
Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–510.
Paugh BS, et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 2013;73(20):6219–29.
Bax DA, et al. EGFRvIII deletion mutations in pediatric high-grade glioma and response to targeted therapy in pediatric glioma cell lines. Clin Cancer Res. 2009;15(18):5753–61.
Gallia GL, et al. PIK3CA gene mutations in pediatric and adult glioblastoma multiforme. Mol Cancer Res. 2006;4(10):709–14.
Grill J, et al. Critical oncogenic mutations in newly diagnosed pediatric diffuse intrinsic pontine glioma. Pediatr Blood Cancer. 2012;58(4):489–91.
Bettegowda C, et al. Exomic sequencing of four rare central nervous system tumor types. Oncotarget. 2013;4(4):572–83.
Kleinschmidt-DeMasters BK, et al. Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am J Surg Pathol. 2013;37(5):685–98.
Knudsen ES, Wang JY. Targeting the RB-pathway in cancer therapy. Clin Cancer Res. 2010;16(4):1094–9.
Barton KL, et al. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PLoS One. 2013;8(10):e77639.
Vazquez A, et al. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7(12):979–87.
Sung T, et al. Preferential inactivation of the p53 tumor suppressor pathway and lack of EGFR amplification distinguish de novo high grade pediatric astrocytomas from de novo adult astrocytomas. Brain Pathol. 2000;10(2):249–59.
Stommel JM, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318(5848):287–90.
Fouladi M, et al. A molecular biology and phase II trial of lapatinib in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Neurooncol. 2013;114(2):173–9.
Broniscer A, et al. Phase I and pharmacokinetic studies of erlotinib administered concurrently with radiotherapy for children, adolescents, and young adults with high-grade glioma. Clin Cancer Res. 2009;15(2):701–7.
Geoerger B, et al. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur J Cancer. 2012;48(2):253–62.
Geoerger B, et al. Innovative therapies for children with cancer pediatric phase I study of erlotinib in brainstem glioma and relapsing/refractory brain tumors. Neuro Oncol. 2011;13(1):109–18.
Pollack IF, et al. A phase II study of gefitinib and irradiation in children with newly diagnosed brainstem gliomas: a report from the Pediatric Brain Tumor Consortium. Neuro Oncol. 2011;13(3):290–7.
Pollack IF, et al. Phase I trial of imatinib in children with newly diagnosed brainstem and recurrent malignant gliomas: a Pediatric Brain Tumor Consortium report. Neuro Oncol. 2007;9(2):145–60.
Broniscer A, et al. Phase I study of vandetanib during and after radiotherapy in children with diffuse intrinsic pontine glioma. J Clin Oncol. 2010;28(31):4762–8.
Fouladi M, et al. Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol. 2011;29(26):3529–34.
Gururangan S, et al. Lack of efficacy of bevacizumab plus irinotecan in children with recurrent malignant glioma and diffuse brainstem glioma: a Pediatric Brain Tumor Consortium study. J Clin Oncol. 2010;28(18):3069–75.
Haas-Kogan DA, et al. Phase II trial of tipifarnib and radiation in children with newly diagnosed diffuse intrinsic pontine gliomas. Neuro Oncol. 2011;13(3):298–306.
MacDonald TJ, et al. Phase II study of cilengitide in the treatment of refractory or relapsed high-grade gliomas in children: a report from the Children’s Oncology Group. Neuro Oncol. 2013;15(10):1438–44.
Broniscer A, et al. Phase I trial, pharmacokinetics, and pharmacodynamics of vandetanib and dasatinib in children with newly diagnosed diffuse intrinsic pontine glioma. Clin Cancer Res. 2013;19(11):3050–8.
Schroeder KM, Hoeman CM, Becher OJ. Children are not just little adults: recent advances in understanding of diffuse intrinsic pontine glioma biology. Pediatr Res. 2014;75(1–2):205–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Becher, O.J., Barton, K.L., Halvorson, K.G., McLendon, R. (2015). Pediatric High-Grade Gliomas and DIPG. In: Karajannis, M., Zagzag, D. (eds) Molecular Pathology of Nervous System Tumors. Molecular Pathology Library, vol 8. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1830-0_7
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1830-0_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1829-4
Online ISBN: 978-1-4939-1830-0
eBook Packages: MedicineMedicine (R0)