
Abstract
Gastroenteropancreatic neuroendocrine tumors (GEP-NETs) are rare neoplasms that require a multidisciplinary approach for an optimal management. The lack of effectiveness of traditional DNA-damaging agents has led to the exploration of new targeted drugs in order to exploit phenotypical features of GEP-NET therapy. However, due to the orphan setting of these tumors, deeper characterization of molecular features and pathways that characterize cell growth, apoptosis, angiogenesis, and invasion are lacking, particularly genetic mutations or epigenetic alterations that generate oncogenic dependency or even addiction. The PI3K-AKT-mTOR pathway has been implicated as having a crucial role in GEP-NETs not only due to the overexpression of several growth factors and their receptors that finally activate this axis but also hereditary syndromes with constitutive activation of the mTOR pathway with high incidence of GEP-NETs. In this article, we aim to review the recent development of the main molecules that target mTOR complex and have showed promising activity in the treatment of GEPNETs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Gastroenteropancreatic neuroendocrine tumors (GEP-NETs), which fall into two major categories, pancreatic neuroendocrine tumors (pNETs) and carcinoid tumors, are characterized by being remarkably vascular and expressing several growth factors, including vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), insulin-like growth factor 1 (IGF-1), basic fibroblast growth factor, and transforming growth factor-α and -β [1–5]. In addition, several receptors to these growth factors have also been detected, including stem cell factor receptor (c-KIT), epidermal growth factor receptors (EGFR), VEGF receptors-2 and -3, IGF receptors (IGF-R), and PDGF receptors [4, 6, 7]. The elevated expression of some of these factors has been associated with poor prognosis and decreased progression-free survival [2, 3] as well as with tumor growth, aggressiveness, and disease extent [3] in patients with GEP-NETs.
Another important feature of GEP-NETs is that they exhibit limited susceptibility to traditional cytotoxic agents, and when they do respond, improvements are slow to become evident [8]. This is probably because GEP-NETs usually have a low mitotic rate, rendering DNA-damaging cytotoxics ineffective; also because of their particular biologic properties, such as the presence of gene products related to chemoresistance. In this regard, a recent study by O’Toole et al. assessed some of the factors associated with poor prognosis and low response to cytotoxics in 60 treatment-naïve patients receiving chemotherapy (n = 46) or chemoembolization (n = 14) [9]. In the study, patients were treated with systemic chemotherapy using streptozotocin, doxorubicin, 5-fluorouracil and/or etoposide/cisplatin, and chemoembolization was performed using streptozotocin or doxorubicin. Results showed that expression of Ki-67, CA9, Akt, hypoxia-inducible factor-1, p53, and the DNA mismatch repair protein Mlh1 (mlh1), as well as tumor grade and tumor differentiation, were associated with overall survival in the entire cohort. In addition, expression of Akt was significantly associated with a non-response to cytotoxic therapy (p = 0.04), and expression of phosphatase and tensin homolog deleted from chromosome 10 (PTEN) and hLMH1 was associated with response to treatment in patients who were only receiving systemic chemotherapy (p = 0.03).
Currently, the medical management of functioning pNETs almost invariably involves the use of somatostatin analogs to control many of the symptoms produced by the exaggerated release of neuropeptides, such as flushing and diarrhea. In patients who develop resistance to somatostatin analogs, addition of interferon (INF)-α may be considered [10]. However, the antiproliferative effects of INF-α are not impressive, ranging from 10% to 15% according to different studies [11–13]. Although neither agent has been shown to induce tumor regression, a recent double-blind, prospective, randomized, controlled study demonstrated that octreotide long-acting release (LAR) significantly lengthened time to tumor progression compared to placebo in patients with metastatic midgut NETs [14], an effect that had not been shown in previous studies of somatostatin analogs.
Treatment options for advanced GEP-NETs are limited and largely rely on the use of somatostatin analogs for symptomatic control and chemotherapy in patients with pNETs. Combination regimens containing streptozoicin, dacarbazine, epirubicin, lomustine, cisplatin, doxorubicin, etoposide, and 5-fluorouracil have shown the best efficacy, with response rates ranging from 20% to 60%, and a median overall survival ranging between 15 and 26 months [15–17]. Unfortunately, low-proliferating, well-differentiated GEP-NETs are virtually insensitive to cytotoxic therapy. Upon disease progression, therapeutic options include hepatic artery chemoembolization and radiofrequency.
Surgery is the mainstay of treatment for localized tumors. If detected early, which occurs rarely, surgery can be curative, yielding survival rates between 80% and 100% [18]. In metastatic disease, surgery may be performed to reduce tumor mass, before or concomitantly with systemic therapy, and to palliate symptoms without cytoreductive intent [18, 19]. Nonetheless, most patients with advanced disease will die from their disease.
In view of the above, there is an evident need to identify and develop novel therapies with proven impact on survival. In this regard, targeted therapies directed against VEGF or components of its downstream signaling pathway have become of particular interest. Multikinase inhibitors, such as sunitinib, the VEGF inhibitor bevacizumab, and inhibitors of the mammalian target of rapamycin (mTOR), such as everolimus, are currently the most tested targeted therapies in phase II and III trials, either alone or in combination chemotherapy [20].
2 mTOR signaling pathway
mTOR is an intracellular serine/threonine kinase found virtually in all mammalian cells. Under physiologic conditions, mTOR participates in several processes, including protein translation, cell growth, proliferation, survival, metabolism, and autophagy [21]. However, hyperactive mTOR signaling has also been shown to be a key participant in the development, growth, and proliferation of many types of human cancers [22]. Therefore, components of mTOR signaling pathway currently constitute targets for the development of anticancer therapies.
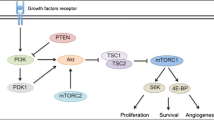
In cells, mTOR acts as the catalytic subunit of two functionally distinct complexes, named mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). This latter complex is considered the main effector of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway, which is dysregulated in most human cancers [23]. mTORC1 consists of mTOR, the target of rapamycin complex subunit LST8, the proline-rich Akt substrate 1, and the regulatory associated protein of mTOR. On the other hand, mTORC2 consists of mTOR, LST8, the target of rapamycin complex 2 subunit MAPKAP1 (SIN1), the proline-rich protein 5 (PRR5, or PROTOR1), and the rapamycin-insensitive companion of mTOR [22].
Cellular processes coordinated by mTOR signaling are regulated by positive and negative regulators, as well as by hormones, nutrients (amino acids, glucose), cellular energy status, and stress conditions. Positive regulators include growth factors, such as IGF-1 and 2, PDGF, and VEGF, and their receptors (IGF-R, VEGFR, PDGRF, EGFR) and associated ligands. Negative regulators of mTOR signaling include the tumor suppressor PTEN, which negatively regulates PIK3 signaling, the tuberous sclerosis complex (TSC) 1 and 2, and the tumor suppressor and protein kinase LKB1 [24]. Activation of mTORC1 ultimately leads to enhanced cell proliferation, survival, and angiogenesis through the selective synthesis of proteins such as G1/S-specific cyclin-D1, Bcl-2, the Bcl2 antagonist of cell death (BAD), and VEGF. mTORC1 also regulates the expression of angiogenic growth factors, such as VEGF and PDGF-β, by phosphorylating hypoxia-inducible factor-1α (HIF-1α) [22, 24, 25]. Activation of mTORC2 results in the phosphorylation of RAC-alpha serine/threonine–protein kinase, and the serine/threonine–protein kinase Sgk1 at the hydrophobic motif. mTORC2 also mediates the hyperactive PI3K activity. Activation of this downstream mTOR pathway regulates cell growth, survival, apoptosis, and proliferation through the activation/inactivation of factors such as Bcl-2, BAD, the apoptosis signal-regulating kinase 1, p53, and the cyclin-dependent kinase inhibitor 1B (p27Kip1) [25, 26].
The molecular pathway of mTOR has been the subject of numerous cancer studies because genetic alterations or increased expression of the components involved in the PI3K/Akt/mTOR pathway have been shown to induce malignant cellular transformation, dysregulation of proliferation, and chemoresistance [23, 27, 28] and also because one of the effects of mTOR activation is the induction of VEGF expression through HIF-1α phosphorylation, which contributes to tumorigenesis and tumor growth [2].
3 Preclinical development of mTOR inhibitors in NETs
Several targets of the PI3K/Akt/mTOR pathway have been identified in GEP-NETs, including an increased expression of IGFs and IGF-R [3, 7]. Using human pancreatic carcinoid tumor cells (BON cells), which express functionally active IGF-1R and secrete IGF-1, it has been shown that IGF-1 is a major autocrine regulator of neuroendocrine secretion and tumor growth [29]. Also, the induction of the raf-1/MEK1 pathway has been demonstrated to block IGF-1-mediated intracellular neuroendocrine hormone regulation, thus representing a viable method to block IGF-1-mediated cellular effects and a potential therapeutic target in carcinoid tumors [30]. Treatment of BON cells with IGF-1 stimulated the release of CgA while intracellular CgA levels were maintained. However, parallel activation of the raf-1/MEK1 pathway reversed the effect of IGF-1 treatment, as reflected by a decrease in intracellular levels of CgA [30].
More recently, Zatelli et al. [31] conducted a study to evaluate the antiproliferative effects of everolimus on bronchial carcinoids (i.e., carcinoid lung tumors), which is an infrequent tumor that originates from endocrine cells dispersed in the respiratory endothelium. Primary cultures of the tumors were treated with everolimus, and cell viability, CgA and VEGF secretion, and somatostatin receptors, mTOR and AKT expression, were assessed. In 67.5% of the samples, everolimus significantly reduced cell viability by approximately 30% (p < 0.05), inhibited p70S6K activity (by 30%), and blocked IGF-1-mediated proliferative effects. As to CgA and VEGF secretion, everolimus induced a significant reduction of 15–20%. Expression of AKT receptors was not different to that of controls, but mTOR expression was significantly higher in the responder group.
Importantly, the identification of components of the mTOR signaling pathway in several human genetic diseases associated with the development of pNETs, has also contributed to the rationale for the development of mTOR inhibitors.
3.1 Tuberous sclerosis complex
TSC is an autosomal dominant disease caused by mutations in the TSCI or TSC2 gene, which encode the protein products hamartin and tuberin, respectively. These two proteins are known inhibitors of the mTOR-mediated signaling to the eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) and the ribosomal protein S6 kinase 1 (S6K1) [32], and critical for cell growth and proliferation. Scattered reports have documented the association of tuberous sclerosis with the occurrence of GEP-NETs, through a process probably involving a dysregulation of mTOR-mediated downstream signaling [33, 34]. According to a systematic review in this area, this association seems more firm in the case of pNETs, particularly insulinomas [35].
3.2 Neurofibromatosis type 1
Neurofibromatosis type 1 (NF-1) is a familial cancer syndrome caused by mutations in the NF-1 gene, which encodes the protein neurofibromin. It belongs to the same family of diseases as TSC, Cowden disease, and Peutz–Jeghers syndrome. Besides functioning as a Ras-GTPase-activating protein [36], neurofibromin was shown to tightly regulate the mTOR pathway in NF-1-deficient primary cells through a mechanism dependent on Ras and PI3K, and mediated by the phosphorylation and inactivation of tuberin by Akt [37].
3.3 Von Hippel–Lindau disease
Von Hippel–Lindau disease (VHL) is a rare, autosomal dominant disease that results from a mutation in the VHL tumor suppressor gene. Patients with VHL are prone to develop pNETs; epidemiological studies report that as many as 17% of patients with this disease develop pNETs [38]. The VHL protein (pVHL) has a key role in the regulation of HIF complex. Under physiologic conditions, pVHL binds to HIF-1α, targeting it for oxygen-dependent polyubiquitination and degradation by proteasomes. However, mutated pVHL is unable to bind to HIF-1α, preventing its degradation. In consequence, excess HIF-1α may interact with HIF-1β, resulting in the formation of a heterodimeric transcription factor that is able to activate the transcription of several hypoxia-inducible genes, including VEGF [39, 40].
3.4 Multiple endocrine neoplasia type 1
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant disease that largely (>90% of the cases) results from mutations in the MENIN gene. Menin, the gene product of MENIN, is mostly a nuclear protein that participates in transcriptional regulation, genome stability, cell division, and proliferation. Patients with this disease develop parathyroid, anterior pituitary, endocrine pancreas, and duodenum tumors, and rarely, NETs and pNETs [41]. However, some reports suggest that the tumorigenesis of sporadic carcinoid tumors and sporadic pNETs is associated with mutations in MENIN based on the fact that approximately 40% exhibit loss of heterozygosity at chromosome 11, and specifically at 11q13, the site of the MENIN gene [42–45].
Finally, the use of gene expression analysis has allowed the identification of potential therapeutic targets and biomarkers to predict patient outcome. In a recent study, TSC2 and PTEN were shown to be downregulated in most primary pNET samples assessed. In addition, their low expression was significantly associated with lower overall and disease-free survival rates. Importantly, expression of FGF13 was significantly associated with the presence of liver metastases and a shorter disease-free survival [46]. Findings of this study further support the role of the mTOR pathway in pNETs and add to the wealth of data that has spurred the clinical development of mTOR inhibitors.
4 Clinical development of mTOR inhibitors in the treatment of NETs
4.1 Temsirolimus
Temsirolimus is an intravenous mTOR inhibitor currently approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMEA) for the treatment of advanced renal cell carcinoma. Temsirolimus binds to the intracellular protein peptidyl-prolyl cis-trans isomerase FKBP1A (FKBP-12), forming a complex that inhibits the activity of mTOR. This effect results in a G1 phase growth arrest, a blockade of its ability to phosphorylate S6K1 and the ribosomal protein S6, and a reduction of HIF-1α, HIF-2α, and VEGF expression [47].
Duran et al. [48] conducted a phase II trial to evaluate the efficacy, safety, and pharmacodynamics of temsirolimus in patients with advanced GEP-NETs, either carcinoid tumors or islet cell carcinoma (i.e., pNETs). Objective tumor response rate, time to progression, overall survival, and adverse events were evaluated. Thirty-six patients (15 patients with islet cell carcinoma and 21 with carcinoid tumors) were treated with weekly intravenous doses of temsirolimus 25 mg.
For the entire cohort, the intention-to-treat analysis showed a response rate of 5.6% (95% confidence interval (CI), 0.6–18.7), a median time to progression of 6 months, and a 1-year overall survival rate of 71.5%. One patient with islet cell carcinoma (6.7%) and one patient with carcinoid tumor (4.8%) achieved partial responses. The treatment was well tolerated. After a median of four cycles delivered per patient, the most frequent adverse events were fatigue (78% of patients), hyperglycemia (69% of patients), and rash/desquamation (64% of patients).
The pharmacodynamic analysis showed that temsirolimus effectively inhibited the phosphorylation of the ribosomal protein S6 (p = 0.02). In addition, higher baseline levels of phosphorylated mTOR were predictive of a better response (p = 0.01). An increased expression of Akt and a decreased expression of phosphorylated mTOR after 2 weeks of treatment were both associated with an increase in time to progression (p = 0.04 and p = 0.05, respectively).
Although the results of this study confirmed temsirolimus’ efficacy in inhibiting mTOR’s downstream signaling pathway, the limited clinical efficacy did not support its use as single agent in patients with GEP-NETs [48].
4.2 Everolimus
In contrast, the clinical development of everolimus for the treatment of patients with GEP-NETs has been more successful. This oral mTOR inhibitor has recently obtained US FDA and EMEA approval for the treatment of patients with advanced renal cell carcinoma after failure of treatment with sunitinib or sorafenib. Similar to temsirolimus, everolimus binds to FKBP-12, forming a complex that induces the inhibition of mTOR kinase activity, a reduction in the activity of mTOR’s downstream effectors S6K1 and 4E-BP1, an inhibition of HIF-1α expression, and a decrease in VEGF expression. In in vivo and in vitro studies, everolimus has been shown to reduce cell proliferation, angiogenesis, and glucose uptake [49]. In human pancreatic BON cells, everolimus exerted a potent dose-dependent inhibition of cell growth involving G0/G1 phase arrest as well as induction of apoptosis [50].
In a phase I pharmacodynamic study of everolimus in patients with advanced solid tumors, including NETs, everolimus induced a dose- and schedule-dependent inhibition of the mTOR pathway, with an almost complete inhibition of phosphorylated ribosomal protein S6 (p < 0.001) and eIF4G (p < 0.001) expression at 10 mg/day and ≥50 mg/week. There was a trend towards significance in the reduction of phosphorylated 4E-BP1 expression (p = 0.058). In addition, an overall increase in Akt phosphorylation (p = 0.006) and in cellular proliferation (p = 0.014) was observed. A clinical benefit was observed in four of the 55 patients (a partial response was observed in one patient, and three had stable disease). Dose-limiting toxicities occurred in five patients, and consisted of grade 3 stomatitis, neutropenia, and hyperglycemia [51]. Based on these results, a dose of 10 mg/day or 50 mg/week was recommended for further development.
Results from two phase II trials by Yao et al. on everolimus in combination with octreotide LAR [52] or alone [53] confirmed the efficacy and safety of everolimus in patients with hard-to-treat GEP-NETs. In the first trial, 60 patients with advanced low- to intermediate-grade GEP-NETs (30 patients with carcinoid tumors and 30 with islet cell carcinomas) were treated with intramuscular octreotide LAR 30 mg every 28 days and oral everolimus, 5 mg/day (patients 1 to 30) or 10 mg/day (patients 31 to 60) every 28 days.
The intention-to-treat analysis revealed a response rate of 20%, and the per protocol analysis showed that 13 (22%) patients achieved partial responses, 42 (70%) patients had stable disease, and five (8%) patients progressed. Median progression-free survival was 13.8 months, and the median overall survival had not been reached at the time of study publication. One-, 2-, and 3-year survival rates were 83%, 81%, and 78%, respectively. Of 37 patients with high chromogranin A levels at study entry, 70% achieved normalization or a reduction of more than 50%. In patients from whom pre- and post-treatment tumor biopsies were available, mean tumor Ki-67 expression (as detected by immunohistochemistry) decreased significantly from 6.7% to 2.1% (p = 0.04). Grade 3/4 adverse events included hypophosphatemia, fatigue, and diarrhea, all of which occurred in 11% of patients. These results confirmed that treatment with everolimus at 5 or 10 mg/day in combination with octreotide LAR was well tolerated by patients with GEP-NETs and supported its antitumor activity in advanced low- to intermediate-grade tumors (Table 1) [52].
In the next phase II trial known as the “RAD001 In Advanced Neuroendocrine Tumors” (RADIANT)-1 trial, patients with advanced islet cell carcinoma who had progressed after cytotoxic chemotherapy were stratified according to prior octreotide therapy into two strata [53]. In stratum 1, 115 patients were treated with everolimus 10 mg/day; in stratum 2, 45 patients were treated with everolimus 10 mg/day plus octreotide LAR ≤ 30 mg every 28 days. The primary end point was to determine response rates in patients in stratum 1. Secondary end points were to determine duration of response, safety, progression-free survival, and pharmacokinetics in both strata, and response rates in patients in stratum 2.
In an intention-to-treat analysis, in stratum 1, 11 (9.6%) patients showed partial response and 78 (67.8%) had stable disease, yielding a clinical benefit of 77.4%. In stratum 2, two (4.4%) patients achieved partial response and 36 (80.0%) patients had stable disease, yielding a clinical benefit of 84.4%. Median progression-free survival was 9.7 months in patients in stratum 1 and 16.7 months in patients in stratum 2. The majority of adverse events were mild to moderate and were consistent with those previously described for everolimus. Once again, this study, carried out in a larger sample of patients with advanced or metastatic GEP-NETs, supported the antitumor activity and safety of everolimus 10 mg/day with or without concomitant administration of octreotide LAR, as measured by response rates and progression-free survival (Table 1) [53].
Findings from these two trials led to the development of two pivotal phase III randomized trials, the RADIANT-2 and RADIANT-3 trials. Very recently, preliminary results of the RADIANT-2 trial were presented at the 35th European Society for Medical Oncology (ESMO) Congress (Milan, Italy, October 8–12, 2010) [54]. In this trial, the efficacy and safety of everolimus plus octreotide LAR were compared to that of placebo plus octreotide LAR in 429 patients with advanced carcinoid tumors. Eligible patients were randomized 1:1 to receive either oral everolimus 10 mg/day plus octreotide LAR 30 mg every 28 days or placebo plus octreotide LAR. The primary end point was progression-free survival. Secondary end points included safety, overall response rate, and overall survival.
Although the study did not meet its primary end point based on central radiologic review of data (p = 0.026 versus p = 0.0246 predefined), results showed that everolimus plus octreotide LAR significantly improved progression-free survival by 5.1 months (hazard ratio = 0.77; 95% CI, 0.59–1.00; p = 0.026) when compared with placebo. After adjusting for imbalances in baseline characteristics between the two treatment arms, and informative censoring in central review [54], the results also showed that everolimus plus octreotide LAR significantly reduced the risk of disease progression by 40% (hazard ratio = 0.60; 95% CI, 0.44–0.84; p = 0.0014) when compared to octreotide LAR alone (Table 1) [54].
In the RADIANT-3 phase III trial, the efficacy and safety of everolimus plus best supportive care (BSC) were compared to placebo plus BSC in patients with advanced pNETs. Four hundred ten eligible patients were randomized 1:1 to receive either daily oral everolimus 10 mg/day or placebo. Preliminary results, which were also presented at the 35th ESMO Congress showed that, compared to placebo, progression-free survival more than doubled in patients receiving everolimus, from 4.6 to 11.0 months (hazard ratio = 0.35; 95% CI, 0.27–0.45; p < 0.0001). Stomatitis, anemia, and hyperglycemia were the most common grade 3/4 adverse events, observed in 6.9%, 6.0%, and 5.0% of patients, respectively (Table 1) [55].
Results from the RADIANT trials, the largest in patients with advanced NETs, are important and could be pointing towards a change in the treatment paradigm of these difficult-to-treat tumors.
5 Conclusions
Treatment options for patients with GEP-NETs are limited, particularly for patients with asymptomatic carcinoid tumors with low-proliferating and well-differentiated tumors and patients in whom the disease has progressed despite treatment with chemotherapy or somatostatin analogs.
mTOR is a key regulator of cell growth, proliferation, and survival, both in physiologic and pathologic conditions. Hyperactivity of mTOR signaling has been shown to be critically involved in the development and proliferation of many cancers. Thus, inhibition of this pathway constitutes an attractive target for the development of biologic agents. To date, the mTOR inhibitor everolimus has shown the most promising results. The efficacy and safety of everolimus for the treatment of patients with advanced low- to intermediate-grade GEP-NETs, either alone or in combination with octreotide LAR, is supported by recent results from phase II and III trials (RADIANT trials). Results from these studies are expected to introduce an important change in the management of these problematic tumors. Combinations of everolimus plus other targeted therapies, such as bevacizumab or temozolomide, are currently being tested in phase I/II trials.
References
Hansel, D. E., Rahman, A., Hermans, J., et al. (2003). Liver metastases arising from well-differentiated pancreatic endocrine neoplasms demonstrate increased VEGF-C expression. Modern Pathology, 16(7), 652–659.
Zhang, J., Jia, Z., Li, Q., et al. (2007). Elevated expression of vascular endothelial growth factor correlates with increased angiogenesis and decreased progression-free survival among patients with low-grade neuroendocrine tumors. Cancer, 109(8), 1478–1486.
Furukawa, M., Raffeld, M., Mateo, C., et al. (2005). Increased expression of insulin-like growth factor I and/or its receptor in gastrinomas is associated with low curability, increased growth, and development of metastases. Clinical Cancer Research, 11(9), 3233–3242.
Chaudhry, A., Papanicolaou, V., Oberg, K., et al. (1992). Expression of platelet-derived growth factor and its receptors in neuroendocrine tumors of the digestive system. Cancer Research, 52(4), 1006–1012.
Nilsson, O., Wangberg, B., Kolby, L., et al. (1995). Expression of transforming growth factor alpha and its receptor in human neuroendocrine tumours. International Journal of Cancer, 60(5), 645–651.
Fjällskog, M. L., Lejonklou, M. H., Oberg, K. E., et al. (2003). Expression of molecular targets for tyrosine kinase receptor antagonists in malignant endocrine pancreatic tumors. Clinical Cancer Research, 9(4), 1469–1473.
Wulbrand, U., Remmert, G., Zofel, P., et al. (2000). mRNA expression patterns of insulin-like growth factor system components in human neuroendocrine tumours. European Journal of Clinical Investigation, 30(8), 729–739.
Kaltsas, G. A., Besser, G. M., & Grossman, A. B. (2004). The diagnosis and medical management of advanced neuroendocrine tumors. Endocrine Reviews, 25(3), 458–511.
O’Toole, D., Couvelard, A., Rebours, V., et al. (2010). Molecular markers associated with response to chemotherapy in gastro-entero-pancreatic neuroendocrine tumors. Endocrine-Related Cancer, 17(4), 847–856.
Kolby, L., Persson, G., Franzen, S., et al. (2003). Randomized clinical trial of the effect of interferon alpha on survival in patients with disseminated midgut carcinoid tumours. The British Journal of Surgery, 90(6), 687–693.
Moertel, C. G., Rubin, J., & Kvols, L. K. (1989). Therapy of metastatic carcinoid tumor and the malignant carcinoid syndrome with recombinant leukocyte A interferon. Journal of Clinical Oncology, 7(7), 865–868.
Öberg, K., Norheim, I., Lind, E., et al. (1986). Treatment of malignant carcinoid tumors with human leukocyte interferon: Long-term results. Cancer Treatment Reports, 70(11), 1297–1304.
Dirix, L. Y., Vermeulen, P. B., Fierens, H., et al. (1996). Long-term results of continuous treatment with recombinant interferon-alpha in patients with metastatic carcinoid tumors—an antiangiogenic effect? Anti-Cancer Drugs, 7(2), 175–181.
Rinke, A., Muller, H. H., Schade-Brittinger, C., et al. (2009). Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. Journal of Clinical Oncology, 27(28), 4656–4663.
Moertel, C. G., Lefkopoulo, M., Lipsitz, S., et al. (1992). Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. The New England Journal of Medicine, 326(8), 519–523.
Kouvaraki, M. A., Ajani, J. A., Hoff, P., et al. (2004). Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. Journal of Clinical Oncology, 22(23), 4762–4771.
Sun, W., Lipsitz, S., Catalano, P., et al. (2005). Phase II/III study of doxorubicin with fluorouracil compared with streptozocin with fluorouracil or dacarbazine in the treatment of advanced carcinoid tumors: Eastern Cooperative Oncology Group Study E1281. Journal of Clinical Oncology, 23(22), 4897–4904.
Öberg, K., & Jelic, S. (2009). Neuroendocrine gastroenteropancreatic tumors: ESMO clinical recommendation for diagnosis, treatment and follow-up. Annals of Oncology, 20(Suppl 4), 150–153.
Öberg, K., Kvols, L., Caplin, M., et al. (2004). Consensus report on the use of somatostatin analogs for the management of neuroendocrine tumors of the gastroenteropancreatic system. Annals of Oncology, 15(6), 966–973.
Capdevila, J., & Salazar, R. (2009). Molecular targeted therapies in the treatment of gastroenteropancreatic neuroendocrine tumors. Target Oncology, 4(4), 287–296.
Wullschleger, S., Loewith, R., & Hall, M. N. (2006). TOR signaling in growth and metabolism. Cell, 124(3), 471–484.
Meric-Bernstam, F., & Gonzalez-Angulo, A. M. (2009). Targeting the mTOR signaling network for cancer therapy. Journal of Clinical Oncology, 27(13), 2278–2287.
Arcaro, A., & Guerreiro, A. S. (2007). The phosphoinositide 3-kinase pathway in human cancer: genetic alterations and therapeutic implications. Current Genomics, 8(5), 271–306.
Yuan, R., Kay, A., Berg, W. J., et al. (2009). Targeting tumorigenesis: development and use of mTOR inhibitors in cancer therapy. Journal of Hematology and Oncology, 2, 45.
Vignot, S., Faivre, S., Aguirre, D., et al. (2005). mTOR-targeted therapy of cancer with rapamycin derivatives. Annals of Oncology, 16(4), 525–537.
Franke, T. F., Hornik, C. P., Segev, L., et al. (2003). PI3K/Akt and apoptosis: size matters. Oncogene, 22(56), 8983–8998.
Paez, J., & Sellers, W. R. (2003). PI3K/PTEN/AKT pathway. A critical mediator of oncogenic signaling. Cancer Treatment and Research, 115, 145–167.
LoPiccolo, J., Blumenthal, G. M., Bernstein, W. B., et al. (2008). Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resistance Updates, 11(1–2), 32–50.
von Wichert, G., Jehle, P. M., Hoeflich, A., et al. (2000). Insulin-like growth factor-I is an autocrine regulator of chromogranin A secretion and growth in human neuroendocrine tumor cells. Cancer Research, 60(16), 4573–4581.
Van Gompel, J. J., & Chen, H. (2004). Insulin-like growth factor 1 signaling in human gastrointestinal carcinoid tumor cells. Surgery, 136(6), 1297–1302.
Zatelli, M. C., Minoia, M., Martini, C., et al. (2010). Everolimus as a new potential antiproliferative agent in aggressive human bronchial carcinoids. Endocrine-Related Cancer, 17(3), 719–729.
Tee, A. R., Fingar, D. C., Manning, B. D., et al. (2002). Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proceedings of the National Academy of Sciences of the United States of America, 99(21), 13571–13576.
Verhoef, S., van Diemen-Steenvoorde, R., Akkersdijk, W. L., et al. (1999). Malignant pancreatic tumour within the spectrum of tuberous sclerosis complex in childhood. European Journal of Pediatrics, 158(4), 284–287.
Eledrisi, M. S., Stuart, C. A., & Alshanti, M. (2002). Insulinoma in a patient with tuberous sclerosis: Is there an association? Endocrine Practice, 8(2), 109–112.
Dworakowska, D., & Grossman, A. B. (2009). Are neuroendocrine tumours a feature of tuberous sclerosis? A systematic review. Endocrine-Related Cancer, 16(1), 45–58.
Hattori, S., Maekawa, M., & Nakamura, S. (1992). Identification of neurofibromatosis type I gene product as an insoluble GTPase-activating protein toward ras p21. Oncogene, 7(3), 481–485.
Johannessen, C. M., Reczek, E. E., James, M. F., et al. (2005). The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proceedings of the National Academy of Sciences of the United States of America, 102(24), 8573–8578.
Blansfield, J. A., Choyke, L., Morita, S. Y., et al. (2007). Clinical, genetic and radiographic analysis of 108 patients with von Hippel–Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs). Surgery, 142(6), 814–818. discussion 818 e1–2.
Forsythe, J. A., Jiang, B. H., Iyer, N. V., et al. (1996). Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Molecular and Cellular Biology, 16(9), 4604–4613.
Woodward, E. R., & Maher, E. R. (2006). Von Hippel–Lindau disease and endocrine tumour susceptibility. Endocrine-Related Cancer, 13(2), 415–425.
Thakker, R. V. (2010). Multiple endocrine neoplasia type 1 (MEN1). Best Practice & Research. Clinical Endocrinology & Metabolism, 24(3), 355–370.
Debelenko, L. V., Zhuang, Z., Emmert-Buck, M. R., et al. (1997). Allelic deletions on chromosome 11q13 in multiple endocrine neoplasia type 1-associated and sporadic gastrinomas and pancreatic endocrine tumors. Cancer Research, 57(11), 2238–2243.
Jakobovitz, O., Nass, D., DeMarco, L., et al. (1996). Carcinoid tumors frequently display genetic abnormalities involving chromosome 11. The Journal of Clinical Endocrinology and Metabolism, 81(9), 3164–3167.
Öberg, K. (2009). Genetics and molecular pathology of neuroendocrine gastrointestinal and pancreatic tumors (gastroenteropancreatic neuroendocrine tumors). Current Opinion in Endocrinology, Diabetes and Obesity, 16(1), 72–78.
Wang, E. H., Ebrahimi, S. A., Wu, A. Y., et al. (1998). Mutation of the MENIN gene in sporadic pancreatic endocrine tumors. Cancer Research, 58(19), 4417–4420.
Missiaglia, E., Dalai, I., Barbi, S., et al. (2010). Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. Journal of Clinical Oncology, 28(2), 245–255.
Temsirolimus (Torisel®) (2010). Full prescribing information. http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022088s008lbl.pdf. Accessed 13 Oct 2010.
Durán, I., Kortmansky, J., Singh, D., et al. (2006). A phase II clinical and pharmacodynamic study of temsirolimus in advanced neuroendocrine carcinomas. British Journal of Cancer, 95(9), 1148–1154.
Everolimus (Afinitor®) (2010). Full prescribing information. http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022334s004lbl.pdf. Accessed 13 Oct 2010.
Zitzmann, K., De Toni, E. N., Brand, S., et al. (2007). The novel mTOR inhibitor RAD001 (everolimus) induces antiproliferative effects in human pancreatic neuroendocrine tumor cells. Neuroendocrinology, 85(1), 54–60.
Tabernero, J., Rojo, F., Calvo, E., et al. (2008). Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. Journal of Clinical Oncology, 26(10), 1603–1610.
Yao, J. C., Phan, A. T., Chang, D. Z., et al. (2008). Efficacy of RAD001 (Everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: results of a phase II study. Journal of Clinical Oncology, 26(26), 4311–4318.
Yao, J. C., Lombard-Bohas, C., Baudin, E., et al. (2010). Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: a phase II trial. Journal of Clinical Oncology, 28(1), 69–76.
Pavel, M., Hainsworth, J.D., Baudin, E., et al. (2010). A randomized, double-blind, placebo-controlled, multicenter phase III trial of everolimus plus octreotide LAR vs ersus placebo plus octreotide LAR in patients with advanced neuroendocrine tumors (NET) (RADIANT-2). In: 35th European Medical Oncology Society; 2010; Milan, Italy.
Yao, J.C., Shah, M.H., Ito, T., et al. (2010). A randomized, double-blind, placebo-controlled, multicenter phase III trial of everolimus in patients with advanced pancreatic neuroendocrine tumors (PNET) (RADIANT-3). In: 35th European Society Medical Oncology; 2010; Milan, Italy
Acknowledgements
The author acknowledges Dr. Ximena Alvira from HealthCo, SL (Madrid, Spain) for her assistance in the preparation of this manuscript and Pfizer Spain for the financial support of medical writing services.
Conflicts of interest
The authors declare that they do not have any conflict of interest that may inappropriately influence this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Capdevila, J., Salazar, R., Halperín, I. et al. Innovations therapy: mammalian target of rapamycin (mTOR) inhibitors for the treatment of neuroendocrine tumors. Cancer Metastasis Rev 30 (Suppl 1), 27–34 (2011). https://doi.org/10.1007/s10555-011-9290-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-011-9290-3