
Abstract
There have been major developments in our understanding of the histopathological classification, genetics, molecular signaling pathways, and treatment of neuroendocrine tumors (NETs) over the last decade. The phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway is a promising target for well-differentiated NETs. The recent success of everolimus, an inhibitor of the mammalian target of rapamycin, is proof of principle that targeting this pathway will lead to improved outcomes in these patients. Novel therapies targeting angiogenesis, such as bevacizumab and sunitinib, are showing promise in NETs by improving progression-free survival alone or in combination with mTOR inhibitors. There are an unprecedented number of ongoing clinical trials of innovative treatments for this disease, and the development of combination therapy will lead to better therapeutic outcomes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Neuroendocrine tumors (NETs)
- Pancreatic neuroendocrine tumors (pNETs)
- PI3K
- Akt
- mTOR inhibitor
- VEGF
- Everolimus
- Sunitinib
- Bevacizumab
- IGF
5.1 Introduction
Neuroendocrine tumors (NETs) are a heterogeneous group of tumors that are classified based on morphological, functional, and clinical features. They are all epithelial tumors with neuroendocrine differentiation and can arise from multiple sites. NETs are classified as functional (10–30 %) or nonfunctional (50–80 %) based on their production of specific hormones such as insulin, gastrin, glucagon, and somatostatin [1, 2]. Since pancreatic NETs (pNETs) are uniquely responsive to therapy, they are often considered separately from NETs of other primary sites (sometimes called “carcinoid”). NETs are described by primary organ site and can also be grouped according to their presumed embryonic origin, as foregut, midgut, or hindgut. As such, the nomenclature of neuroendocrine tumors is complicated by its variations in origination and multiple classification systems.
The incidence of NETs has been increasing over the last several decades. Based on the Surveillance, Epidemiology, and End Results (SEER) database, there are about 5.25 new cases per 100,000 in 2004, and there have been an overall increased incidence over time for NETs of all gastrointestinal sites [3, 4]. Approximately 64 % of all NETs originate in the gastrointestinal tract [5] and 6 % arise in the pancreas [3]. The pathogenesis of the disease is not well understood; however, some pancreatic NETs are associated with inherited genetic syndromes, including multiple endocrine neoplasia type 1 (MEN1), von Hippel-Lindau disease, neurofibromatosis, and tuberous sclerosis. Nevertheless, the majority of NETs occur sporadically [6].
Appropriately, the medical treatments for NETs are as varied as their biology. Several approaches are available including somatostatin analogs, peptide receptors radionuclide therapy (PRRT), and systemic chemotherapy. Factors considered when choosing therapies include tumor grade, proliferative index, performance status, and site of origin. Despite these therapeutic tools, the majority of advanced NETs will progress despite optimal therapy, and those with a high proliferative index have a poor prognosis.
For non-pancreatic well-differentiated NETs, traditional cytotoxic agents have limited effectiveness due to their lower proliferative index and other genetic properties related to chemoresistance [7]. Currently, the mainstay of treatment for midgut NETs is the somatostatin analog octreotide or octreotide long-acting release (LAR) which results in palliation of symptoms and improves quality of life [8]. In 2009, this approach was validated; the PROMID study demonstrated that octreotide LAR significantly increases progression-free survival (PFS) from 6 to 14 months in patients with both functionally active and inactive tumors of metastatic midgut NET (jejunum, ileum, appendix, and proximal colon) [9]. Once disease progresses, management options include hepatic artery embolization therapies, radiofrequency ablation, or metastasis resection to reduce tumor burden.
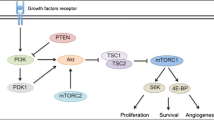
Recent advances in our understanding of the biological features and molecular signaling pathways underlying the progression of NETs have led to the development of novel targeted therapies. In 2011, two new systemic agents: sunitinib, a vascular endothelial growth factor receptor tyrosine kinase inhibitor and everolimus, a mTOR inhibitor, were approved for the treatment of pNETs. These treatments exploit the inherent vascularity and expression of multiple growth factors associated with NETs. Inhibition of PI3K/Akt/mTOR pathway, one of the most important pathways implicated in the pathogenesis of NETs, has improved outcomes and provided new approaches to the treatment of this disease [10]. The goal of this chapter is to review importance of the PI3K/Akt/mTOR pathway in NETs and the development of targeted strategies for this pathway.
5.2 Role of the PI3K/Akt/mTOR Pathway in NET
The recent success of everolimus, an inhibitor of the mammalian target of rapamycin, is proof of principle that the phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway is important to NET tumorigenesis and progression. Gene expression profiling and tumor sequencing studies over the past two decades also confirm the importance of the PI3K/Akt/mTOR pathway to the pathogenesis of NETs (Table 5.1). Alterations in this pathway identified in neuroendocrine tumors include: overexpression of growth factors and receptors, activating mutations in oncogenes, and mutations in tumor suppressor genes. There is substantial and accumulating evidence, both in vitro and in vivo, that mTOR plays an important role in the growth of NETs, particularly pNETs [15, 16].
The PI3K/Akt/mTOR pathway plays an important role in cellular proliferation, growth, and metabolism. This signaling pathway is extensively detailed in another chapter and only a cursory description will be given here. The PI3K family of lipid kinase phosphorylate and the 3′-hydroxyl group of phosphoinositides are composed of three classes (I-III) with distinct lipid products, substrate specificity, and functionality. PI3K and Akt are upstream from the mTOR complexes. The activated PI3K triggers the conversion of phosphatidylinositol-4,5-diphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 then promotes the activation of Akt, also known as protein kinase B, a serine/threonine kinase and is a key regulator of PI3K and mammalian target of rapamycin (mTOR) signaling. Activated Akt stimulates mTOR complex 1 to elicit multiple cellular processes and is an important catalyst of malignant progression and chemoresistance.
mTOR is a serine/threonine kinase and is the downstream effector of the PI3K-activated signaling pathways. It promotes protein synthesis and cell growth during nutrient-rich periods and functions as a sensor of nutritional or metabolic stress during cell development [17]. mTOR regulates apoptosis, proliferation, and cell growth and also modulates mRNA-translation of proteins necessary for cell cycle progression from G1- to S-phase, including E4-binding protein (E4-BP1) and p70 kinase [18]. It represents an important break point in the proliferation and differentiation of tumor cells and is critical for regulating cell proliferation, angiogenesis, and metabolism.
There are two complexes that comprise mTOR, mTOR complex-1 (mTORC1), and mTOR complex-2 (mTORC2). mTORC1 is composed of mTOR, regulatory-associated protein of mTOR (Raptor), and target of rapamycin complex subunit LST8. mTORC1 regulates cellular transcription and translation via eukaryotic translation initiation factor 4E-binding protein-1 (4EBP-1) and ribosomal S6 kinase-1 (S6K1). mTORC2 consists of mTOR and target of rapamycin complex subunit LST8, rapamycin-insensitive companion of mTOR (rictor), and mitogen-activated protein kinase-associated protein-1. The role of mTORC2 is less well defined, but is known to directly phosphorylate Akt in the PI3K-Akt pathway [14].
Clinical syndromes appear to support the role of the PI3K/Akt/mTOR pathway in NET tumorigenesis. Inherited diseases such as multiple endocrine neoplasia type I (MEN1), tuberous sclerosis complex (TSC), neurofibromatosis type I, and von Hippel-Lindau (VHL) disease are associated with an increased incidence of PNETs. Across these syndromes, mutations in well-defined oncogenes and tumor suppressor genes (TSC2, NF1, and vHL genes) lead to constitutive activation of the PI3K/Akt/mTOR pathway. Alterations in PI3K/Akt/mTOR pathway have also been implicated in sporadic pNETs tumorigenesis justifying its exploitation as a target for rationale therapy [12, 14, 19].
Investigations of the PI3K/Akt/mTOR pathway in NETs reveal an association between its activation and cancer development. In neuroendocrine cell lines, PI3K mutations have been associated with response to mTOR inhibition [20]. Activation and phosphorylation of Akt has also been reported in a majority of neuroendocrine tumors [21, 22]. Phosphorylated Akt is a prognostic marker associated with worse outcomes in gastrointestinal NET [23]. MEN1 gene mutations, the hallmark of MEN syndromes, are associated with Akt activation [24]. These mutations have been identified in 10–35 % of foregut NETs and PNETs, both functional and nonfunctional [25–28]. Preclinical studies have also shown that mTOR and its downstream targets are overexpressed in NETs and associated with a higher proliferative index [29]. In clinical studies, expression of mTOR and its pathway components was predictive of response to temsirolimus [30].
Activation of the PI3K/Akt/mTOR pathway is likely driven by dysregulated tyrosine kinases and signaling by vascular endothelial and insulin growth factors. Studies demonstrate that receptors including PDGFR, EGFR, and c-kit are overexpressed in endocrine tumors [31, 32]. NETs and NET cell lines frequently express both IGFs and the IGF-1R receptor suggesting autocrine and/or paracrine signaling [33, 34]. IGF-1R binding leads to the direct activation of signaling cascades in the MAPK and PI3k kinase pathways [35]. The clinical benefit from somatostatin analogs in insulin growth factor secreting tumors suggests an important interplay in NET tumorigenesis and activation of PI3K/Akt/mTOR pathway [36].
Two key negative regulators of the PI3K/Akt/mTOR pathway are phosphatase and tensin homolog (PTEN) and tuberous sclerosis protein 2 (TSC2). PTEN is a tumor suppressor that negatively regulates the PI3K/Akt/mTOR pathway by converting PIP3 back to PIP2 and reversing PI3K activation. TSC2 is phosphorylated and inhibited by Akt which suppresses mTOR signaling thereby attenuating its negative regulation of the PI3K pathway [37]. Based on tissue microarray gene expression analysis, both tumor suppressor proteins were found to be downregulated in 72 primary pNET samples. Furthermore, low expression of TSC2 and PTEN was significantly associated with more aggressive tumors and with shorter disease-free and overall survival [14]. In NETs, PTEN loss or mutation promotes carcinogenesis and is associated with poor differentiation [22, 23].
Amplified angiogenesis is a distinguishing feature of well-differentiated NETs and may be associated with activation of the PI3K/Akt/mTOR pathway [38]. Activation of the PI3K pathway may also be led by the overexpression of VEGFR1 in the companion vasculature suggesting an interaction between this pathway and angiogenesis [25]. Mutations in the FLT1/VEGFR1 gene have been detected in pNET cell lines [25].
5.3 mTORC 1 Inhibitors and NETs
5.3.1 Temsirolimus
Temsirolimus (CCI-779, Torisel®, Pfizer) was the first mTOR inhibitor developed and identified to have antitumor activity [39]. After years of development, it was recently approved for the treatment of advanced renal cell carcinomas and pancreatic neuroendocrine tumors. Temsirolimus forms a complex by binding to the intracellular protein peptidylprolyl cis-trans isomerase FKBP1A (FKBP-12) that inhibits the activity of mTOR. This subsequently results in a G1-phase growth arrest, blocking its ability to phosphorylate S6K1 and the ribosomal protein S6, a reduction of HIF-1α, and VEGF expression [40]. In patients with advanced NET, a phase II study was conducted to evaluate the safety, efficacy, and pharmacodynamics of temsirolimus. Thirty-six patients with advanced and progressive NETs (21 carcinoids and 15 pNET) received weekly doses of intravenous temsirolimus. There was no difference in the objective response rates between carcinoids (4.8 %) and pNET (6.7 %). The intent-to-treat response rate for the entire cohort was 5.6 % (95 % CI 0.6–18.7 %), median TTP was 6 months, and 1-year PFS was 40.1 %. Two patients achieved partial responses (one patient with pNET and one patient with carcinoid tumor). Overall, the treatment was well tolerated with fatigue (78 %), hyperglycemia (69 %), and rash/desquamation (64 %) being the most common drug-related adverse events of all grades after a median of four cycles delivered per patient [30].
Pharmacodynamic analysis demonstrated that temsirolimus effectively inhibited the PI3K/Akt/mTOR pathway. Phosphorylation of the ribosomal protein S6 was significantly depressed (p = 0.02). Additionally, patients with an increased expression of phosphorylated Akt (p = 0.041) and a decreased expression in phosphorylated mTOR after 2 weeks of treatment were both associated with an increase in time to progression (p = 0.04 and p = 0.05, respectively). Elevated baseline levels of phosphorylated mTOR predicted a better response (p = 0.01). Even though the results of this study revealed temsirolimus value in downregulating mTOR’s downstream signaling, the authors concluded that it has limited clinical efficacy and does not support its use as monotherapy in patients with advanced NETs [30].
The limited benefit but excellent tolerability of this agent lends it to be partnered with additional agents. Preclinical studies suggest enhanced antitumor effects with temsirolimus and VEGF-targeted therapy. Therefore, a phase II study of temsirolimus in combination with bevacizumab, an anti-VEGF-A monoclonal antibody, in advanced, recurrent, or progressive pNETs (NCT01010126), was completed. Of the 56 patients eligible for response assessment, partial responses were seen in 41 % (23 of 56) patients, and 79 % of the patients (44/56) had disease stability at 6 months. Median progression-free survival was 13.2 months and overall survival was 34 months. This combination was very well tolerated with minimal toxicity. A minority of patients developed grade 3 or 4 drug-related adverse events including hypertension (18 %), hyperglycemia (13 %), fatigue (11 %), leukopenia (9 %), headache (9 %), proteinuria (7 %), and hypokalemia (7 %). The ORR of 41 % exceeds that reported to date for monotherapy with any targeted agent in pNET and provides compelling evidence to pursue this combination further [41].
5.3.2 Everolimus
Everolimus, a second-generation mTOR inhibitor, was recently approved for use in pNETs after demonstrating significant improvements in outcomes [25, 42]. Everolimus (40-O-(2 hydroxyethyl) derivative of rapamycin, RAD001, Afinitor®, Novartis) is an oral mTOR inhibitor that selectively inhibits mTORC1 and is absorbed rapidly, achieving peak concentration after 1.8 h and reaching steady state after 7 days [16]. It binds to FKBP-12 in a similar mechanism as temsirolimus, by forming a complex that induces the inhibition of mTOR kinase activity. It reduces the activity of mTOR’s downstream proteins by blocking phosphorylation of 4E-BP1 and inactivating S6K1. It also inhibits expression of HIF-1α and decreases expression of VEGF. Everolimus has been shown to reduce cell proliferation, angiogenesis, and glucose uptake from several in vivo and in vitro studies. In addition, it has demonstrated a potent dose-dependent inhibition of cell growth comprising G0/G1 phase arrest as well as induction of apoptosis in human pancreatic BON cells, a human pancreatic NET cell line exhibiting constitutively activation of PI3K/Akt/mTOR pathway [43]. Everolimus treatment also significantly inhibited cell proliferation in the rat insulinoma NET cell line (INS1) and decreased phosphorylation of all downstream targets of Akt, TSC2, and mTOR [44].
In a phase I study evaluating patients with advanced solid tumors including NETs, everolimus induced a dose and schedule-dependent inhibition of the PI3K/Akt/mTOR pathway. At 10 mg/day and ≥50 mg/week, there was an almost complete inhibition of phosphorylated ribosomal protein S6 (p < 0.001), expression of eIF4G (p < 0.001), and reduction of phosphorylated 4E-BP1 (p = 0.058). Furthermore, there was an overall increase in Akt phosphorylation (p = 0.006) and in cellular proliferation (p = 0.014). A total of four out of the 55 patients reached a clinical benefit (a partial response was observed in one patient, and three had stable disease). The dose-limiting toxicities consisted of grade 3 stomatitis, neutropenia, and hyperglycemia which were seen in five patients [45].
A phase II trial evaluated the activity of everolimus in combination with octreotide. Thirty patients with carcinoid and 30 patients with pNETs were treated with everolimus at 5 or 10 mg/day in combination with octreotide LAR 30 mg every 4 weeks. The intent-to-treat response rate was 20 %. The analysis showed that 13 (22 %) patients achieved partial responses, 42 (70 %) patients had stable disease, and 5 (8 %) patients progressed. The median PFS was 60 weeks. Median overall survival had not been reached; however, 1-, 2-, and 3-year survival rates were 83 %, 81 %, and 78 %, respectively. At study entry, among 37 patients with high chromogranin A levels, 26 patients (70 %) attained normalization or a reduction of more than 50 %. In pre- and posttreatment tumor biopsies, mean tumor Ki-67 expression decreased significantly from 6.7 to 2.1 % (p = 0.04). Overall, compared to patients that received the 5 mg dose, patients that received the 10 mg dose obtained a higher response rate (30 vs. 13 %) and had a prolonged median PFS (72 vs. 50 weeks). The most common toxicity was mild aphthous oral ulceration. Significant toxicities were uncommon and only 11 % of patients developed grade 3/4 hypophosphatemia, fatigue, and diarrhea [46].
Three RADIANT (RAD001 in advanced neuroendocrine tumors) trials were then designed to study efficacy of everolimus in NETs of different origins. These confirmed the value of everolimus in patients with advanced NETs. RADIANT-1 is a second open-label phase II trial in 160 patients with progressive chemotherapy-refractory metastatic pNET. Patients were stratified according to octreotide therapy with the primary endpoint assessing response rates in patients in stratum 1. Stratum 1 comprised of 115 patients treated with everolimus 10 mg daily alone and stratum 2 comprised of 45 patients treated with everolimus 10 mg daily plus octreotide LAR ≤30 mg every 28 days. In stratum 1, 11 patients (9.6 %) showed a partial response, 78 patients (67.8 %) had stable disease, and 16 patients (13.9 %) progressed, resulting in a clinical benefit of 77 %. The mean PFS was 9.7 months and overall survival was 24.9 months. In stratum 2, two patients (4.4 %) achieved partial response, 36 patients (80 %) had stable disease, and no patients with progressive disease, resulting in a clinical benefit of 84 %. The mean PFS was 16.7 months and overall survival was not reached after a follow-up period of over 16 months. This study supports the safety and antitumor activity of everolimus alone or in combination with octreotide in patients with progressive pNETs after failure of prior systemic chemotherapy [47].
Following these encouraging results, two pivotal phase III randomized trials were developed. RADIANT-2 evaluated the combination of everolimus plus octreotide LAR compared to octreotide LAR alone in 429 patients with low- to intermediate-grade advanced NET. Although the study failed to reach its primary endpoint, it demonstrated that everolimus plus octreotide LAR significantly improved PFS by 5.1 months (hazard ratio = 0.77, 95 % CI 0.59–1.00, p = 0.026); the mean PFS was 16.4 months in the everolimus plus octreotide LAR group and 11.3 months in the placebo plus octreotide LAR group. After adjusting for differences in baseline characteristics, everolimus plus octreotide LAR also significantly reduced the risk of disease progression by 40 % (hazard ratio = 0.60, 95 % CI 0.44–0.84, p = 0.0014) when compared to octreotide LAR alone [48].
A subgroup analysis of RADIANT-2 trial has shown that early combination therapy with octreotide might be associated with a better outcome compared to patients on octreotide with everolimus added on later (25.2 vs. 13.6 months). COOPERATE-2 study is an ongoing prospective randomized open-label phase II trial in pNET that aims to address the superiority of combination therapy and evaluates the treatment effect of everolimus with a novel somatostatin analog, pasireotide LAR, in comparison to everolimus monotherapy on PFS in patients with advanced progressive pNET (NCT01374451).
RADIANT-3 is the largest phase III pNET trial to date. This was a landmark double-blinded and placebo-controlled study that evaluated 410 patients with advanced, low-grade, or intermediate-grade pNET who received everolimus 10 mg/day (n = 207) or placebo (n = 203). Everolimus more than doubled progression-free survival (11 months vs. 4.6 months (p < 0.0001)) and was associated with a 65 % reduction in progression or death. Although the objective response rate to everolimus was low at 5 % (2 % in the placebo arm), there was a benefit seen in patients with prolonged stable disease in the everolimus arm (73 % vs. 51 % for everolimus and placebo, respectively). The overall survival was not significantly different between the two groups as more than 70 % of patients randomly assigned to placebo crossed over to the treatment arm after disease progression [1, 42]. There was a twofold increase in adverse events; the most common side effects were hematological, diarrhea, stomatitis, or hyperglycemia, ranging from 3 to 7 %. These side effects were manageable with dose reduction, drug interruption, or both [42]. In subgroup analyses, these benefits extended to patients regardless of ethnicity of history of previous therapies [49].
The approval of everolimus has changed the landscape for patients with advanced well-differentiated pNETs. Although the timing of everolimus in the treatment of advanced pNETs is not yet established, everolimus was equally effective in patients regardless of treatment history. The European Neuroendocrine Tumor Society (ENETS) 2012 determined that everolimus represents a novel therapeutic option in patients with unresectable pNETs after progression following chemotherapy, and in selected cases, everolimus should be considered as first-line therapy [50]. Similarly, the National Comprehensive Cancer Network (NCCN) recommended the use of everolimus as a possible first-line treatment for advanced unresectable well-differentiated pNETs (the NCCN clinical practice guidelines in oncology for neuroendocrine tumors version 1.2012. 2012. www.nccn.org).
Although there is convincing data for everolimus in pNETs, its efficacy in other NET subtypes, such as bronchopulmonary or colonic NET, has not been determined. Preclinical studies suggest that susceptibility to everolimus may depend on site of origin for NETs [51]. The RADIANT-4 trial will investigate the benefit of everolimus versus placebo in patients with advanced nonfunctional neuroendocrine tumor of gastrointestinal or lung origin (NCT01524783).
5.4 Resistance Mechanisms of PI3K/Akt/mTOR Pathway Inhibitors
Current mTOR inhibitors have therapeutic limitations as patients initially respond but will eventually progress despite continuous therapy. Primary and acquired resistance appears to limit the efficacy of targeting the PI3K/Akt/mTOR pathway. Escape mechanisms, abrogation of negative feedback loops, and mutations in targeted pathways can all lead to resistance (Table 5.2).
In NET cell lines, mTOR1 inhibitors produce escape mechanisms in both Erk and Akt pathways [51]. For example, rapamycin activity was associated with increased levels of phospho-Akt and phospho-ERK. Akt and ERK are then able to act in concert with RAS and PI3K thereby activating these pathway [53].
Primary resistance mechanisms, such as preexisting mutations in the targeted pathways, may render many targeted therapies ineffective. In tumors harboring K-Ras or B-Raf mutations, resistance is due to activation of alternative pathways, such as the Erk pathway [20].
There is concern for the use of single-agent everolimus in the treatment of NETs due to the presence of feedback loops and crosstalk that exist within and between PI3K/Akt/mTOR and other signaling pathways. Recent data suggest that the loss of negative feedback loops from inhibition of mTORC1 leads to compensatory activation of PI3K and Akt, which drives resistance via upregulation of mTORC2 [54]. Two well-characterized mTORC1 substrates are eukaryotic translation initiation factor 4E-binding protein-1 and ribosomal S6 kinase-1 (S6K1), both regulating transcription and translation initiation of critical growth genes. However, S6K1 is part of a negative feedback loop on PI3K/Akt signaling via suppression of the insulin receptor substrate-1 (IRS1), which links IGF-1 to the PI3K pathway. mTORC2 is less defined than mTORC1, but is known to mediate Akt phosphorylation on serine-473, which is required for full Akt activity in the PIK3/Akt/mTOR signaling cascade. Normally, activation of S6K through mTORC1 phosphorylation results in phosphorylation of rictor, which prevents mTORC2 activation [55, 56]. If mTORC1/S6K is inhibited, the negative feedback is lost leading to increased mTORC2-mediated phosphorylation and activation of Akt [57]. Thus, inhibition of mTORC1 by everolimus may lead to paradoxical upregulation of Akt. This concern has been confirmed in tumor biopsies from patients treated with mTOR inhibitors [58].
Another potential mechanism of resistance is the loss of a negative feedback loop that normally prevents upstream overstimulation of insulin receptor substrate 1 (IRS1) [53]. mTORC1 activation of S6K causes uncoupling of IGF-1 from the PI3K/Akt pathway. Normally, IGF-1 binds IGFR which in turn phosphorylates substrates IRS1-2 which then suppresses PI3K. mTORC1/S6K inhibition results in the loss of this feedback loop and leads to the upregulation of IRS1 protein and activation of the PI3K/Akt cascade [53, 54, 59]. Accordingly, several approaches to downregulate IGF with somatostatin analogs such as octreotide and pasireotide, or inhibit IGF-1R signaling with a monoclonal antibody, such as cixutumumab (IC-A12) are being developed in combination with everolimus. There is an ongoing phase I study with the combination of cixutumumab, everolimus, and octreotide LAR in patients with advanced NETs (NCT01204476).
5.4.1 Novel Approaches
The PI3K/Akt/mTOR pathway is complex and perturbations can occur at multiple sites. Therefore, there are several potential targets and combinations of therapies compelling for further investigation. The use of PI3K inhibitors, Akt inhibitors, or mTORC1 and mTORC2 inhibitors as single agents or in combination can avert PI3K/Akt/mTOR pathway activation and reactivation [55]. A host of novel inhibitors that target key nodes with the PI3K/Akt/mTOR pathway have shown encouraging results in preclinical studies and are currently in early phase clinical trials.
Inhibitors of Akt either compete with ATP at the active site or bind distally to the catalytic site, inducing a conformational change that prevents ligand binding. Akt inhibition may be expected to abrogate negative feedback loops perpetuated by mTORC2 following mTORC1 inhibition [58]. Agents that inhibit both mTOR complexes may also overcome this problem. Therefore, inhibitors of both mTORC1 and mTORC2 and Akt inhibitors are attractive drug candidates.
Potential PI3K/Akt/mTOR pathway target upstreams of mTOR are the PI3K proteins themselves. Three classes (I–III) of PI3K have been characterized that vary in structure and substrate preference. The class I enzymes are activated directly by cell-surface receptors, and it is the catalytic domain of the class IA PI3K p110 subunits that are the most widely implicated in cancer [56]. Pan-PI3K inhibitors target all four class I p110 isoforms; however, PI3K inhibitors specific for individual class I p110 isoforms may allow for anticancer activity with an improved safety profile. The majority of therapeutic interventions or drugs under investigation are pan-p110 inhibitors, although a number of PI3K-targeted agents with isoform specificity have now been reported [55, 57]. It is of potential clinical significance that dual inhibition of PI3K and mTORC1/2 may be mediated through the shared structural homology between the catalytic domains of the PI3K p110 subunits and mTORC1/2 [60]. Agents that target both PI3K and mTOR will likely lead to improved inhibition of this pathway.
5.4.2 mTORC1 and mTORC2 Inhibitors
CC-223 is currently an experimental dual mTOR kinase inhibitor, inhibiting both TORC1 and TORC2 complexes. In a recent phase I trial, 101 solid tumor subjects were treated with CC-223 dosed at 45 mg once daily in 28 day cycles until disease progression. From the non-pancreatic NET cohort (n = 23), patients with progression within 12 months and receiving ongoing treatment with somatostatin analogs were included in the study. Biomarkers confirmed inhibition of TORC1 and TORC2 by p4EBP1 and pAKT, respectively. In 7 out of 13 subjects (54 %), PET imaging demonstrated a response at day 15 (>25 % change in SUV). All evaluable patients were stable on CC-223, with treatment ongoing up to nine cycles (median 6; range 4–9) (n = 10). Although not prospectively collected, there were six subjects with refractory carcinoid syndrome that reported complete resolution of flushing [5] and improvement in diarrhea [1]. Symptom improvement generally occurred within the first week of dosing and persisted despite dose reduction in five subjects [61].
The most common related adverse events of all grades were stomatitis, diarrhea, fatigue, anorexia, nausea, and rash. In addition, related serious adverse events included one case of transient dehydration/renal insufficiency. CC-223 dose reduction to 30 or 15 mg was required in 57 % of subjects, usually during cycle 1 or 2, but thereafter treatment was well tolerated [61].
These results are from an ongoing phase I/II study to assess the safety and efficacy of CC-223 in patients with advanced tumors (other than pNETs) unresponsive to standard therapies and to determine the appropriate dose and tumor type for later-stage clinical trials (NCT01177397).
5.4.3 HSP 90 Inhibitors
There have been numerous preclinical data supporting the role of novel PI3K/Akt/mTOR pathway inhibitors in NETs, either through direct inhibition of specific pathway proteins or through indirect inhibition of molecular chaperones. The molecular chaperone heat-shock protein 90 (HSP90) is an emerging target for anticancer therapy as it is overexpressed in a number of tumors. The HSP90 inhibitor, IPI-504, has been studied in pNET cells. The potential activity of IPI-504 has shown to inhibit the growth of human insulinoma and pancreatic carcinoid cells by almost 70 %. IPI-504 also has antiproliferative effects by downregulating IGF-1 and several downstream factors of the PI3K/Akt/mTOR. Combination of IPI-504 with mTOR or Akt inhibitors also resulted in increasing antiproliferative effects [62]. This is a promising agent for the treatment of NETs.
5.4.4 Insulin Growth Factor Inhibitors
Using BON cells, it has been shown that increased expression of IGF-1 is a major autocrine regulator of neuroendocrine secretion and tumor growth [34]. IGF-1-mediated PI3K/Akt/mTOR signaling has also been targeted with a monoclonal antibody by ganitumab (AMG-479). AMG 479 is a humanized monoclonal antibody to IGFR-1, preventing the binding of IGF-1 and IGF-2 to IGF-1R [63]. This agent has been shown to inhibit PI3K/Akt/mTOR signaling and enhance the antitumor effects of anti-epidermal growth factor receptor (EGFR)-targeted therapies [64].
A phase II study of AMG 479 in NETs and pNETs has completed enrollment and is currently ongoing (NCT01024387). Interim results were presented at ASCO in 2012. Sixty patients (30 carcinoid, 30 pancreatic NET) were treated with AMG 479 18 mg/kg every 3 weeks and 54 patients were evaluable for response. There were no objective responders by RECIST, 10/27 (37 %) evaluable carcinoid patients and 8/26 (31 %) evaluable pancreatic NET patients experienced disease stability, while 17/27 (63 %) of the carcinoid patients and 15/26 (58 %) of the pancreatic NET patients progressed. Median PFS was 10.5 months for carcinoid patients and 4.2 months for pancreatic NET patients. Treatment was well tolerated and significant toxicities were rare.
5.4.5 Akt Inhibitors
Several preclinical studies suggest that directly inhibiting Akt potently reduces the growth of NETs. Akt is a critical signaling node downstream of PI3K important in tumor cell proliferation, growth, survival, and angiogenesis. A number of small-molecule Akt inhibitors for the different Akt isoforms have been developed. The ATP-competitive Akt inhibitors have varying potencies and specificities and have a higher likelihood of off-target effects. Therefore, allosteric Akt inhibitors have been preferred for clinical studies in patients with pNETs given their increased specificity [65]. MK-2206 is an oral allosteric inhibitor of all Akt isoforms. In a phase I trial performed in 33 patients with solid tumors, two patients with advanced pNET had minor responses, achieving tumor shrinkages of 13.1 and 17.5 %. There was a reduction in phosphorylated serine-473 Akt in all tumor biopsies (p = .015) and suppression of phosphorylated threonine 246 proline-rich Akt substrate 40 assessed in hair follicle samples. Reversible hyperglycemia and increased insulin c-peptide associated with Akt inhibitors are consistent with a class effect for mTOR inhibitors. Drug-related toxicities included skin rash (51.5 %), nausea (36.4 %), pruritus (24.2 %), hyperglycemia (21.2 %), and diarrhea (21.2 %). Overall, MK-2206 was well tolerated with evidence of antitumor activity and Akt signaling blockade [66]. There are currently several combination trials with MK-2206 with either standard chemotherapy (carboplatin, paclitaxel, docetaxel) or targeted agents such erlotinib, or lapatinib (a dual EGFR/human epidermal growth factor receptor 2), ridaforolimus (mTORC1 inhibitor), and AZD6244 (MEK1/2 inhibitor).
The Akt inhibitor triciribine has been studied alone and in combination with other therapeutics and has shown a reduction in the growth of NET cells. In preclinical models and cancer cell lines, triciribine significantly reduced insulinoma (CM) cells by 59 % and neuroendocrine tumor cells (STC-1) by 65 %. Notably, triciribine even at higher doses did not inhibit the BON carcinoid cell line. This cell line is characterized by high expression of PTEN, suggesting the role of PTEN as a possible predictor of sensitivity to triciribine in NETs. The Akt pathway also plays an important role in chemotherapy therapy resistance and response to hypoxia and angiogenesis. A synergistic antiproliferative effect has been seen with combination of triciribine and cytostatic drugs as well as drugs targeting a number of proteins of the PI3K/Akt/mTOR pathway [67].
Perifosine, a pan-Akt inhibitor, inhibits both Akt phosphorylation and cell viability in human pancreatic BON1 and other NET cells. Perifosine also suppressed the phosphorylation of Akt downstream targets and induced apoptosis. Studies of individual Akt isoforms for NET have shown that downregulation of Akt isoforms 1 and 3 suppressed NET cell viability, suggesting a particular role for these isoforms in NET signaling. Akt3 siRNA induced apoptosis, while all three isoform-specific siRNAs impaired BON1 cell invasion. These studies highlight the potential for selective Akt isoform targeting in NETs [68].
5.5 Multi-targeted Approaches
NETs are hypervascular tumors that secrete an enormous amount of VEGF. The activation of mTOR results in the induction of VEGF expression by phosphorylating hypoxia-inducible factor 1α (HIF-1α), which contributes to tumorigenesis and tumor growth [69–71]. The relationship between VEGF expression and mTOR activation has encouraged the study of upstream pathway inhibition with several inhibitors such as octreotide, PI3K, and mTOR inhibitors. Villaume et al. analyzed effects of octreotide, mTOR inhibitor (rapamycin), PI3K inhibitor (LY294002), MEK1 inhibitor, and the p38 inhibitor on VEGF secretion in three murine endocrine cell lines, STC-1, INS-r3, and INS-r9. The authors found that octreotide and rapamycin induced a significant decrease in VEGF production by all three cell lines. The PI3K inhibitor significantly inhibited VEGF production in STC-1 and INS-r3 cells only. There was also a decrease in intracellular levels of VEGF and HIF-1α observed for octreotide, mTOR inhibitor, and PI3K inhibitor. The complex regulation of VEGF synthesis and secretion by the mTOR and PI3K inhibitors is likely mediated by the inhibition of the PI3K/Akt/mTOR pathway. It has also been observed by the decrease in Akt phosphorylation detected in all three cell (STC-1, INS-r3, and INS-r9) lines that octreotide may act through inhibition of the PI3K/Akt/mTOR pathway [72].
In a recently completed phase II study, the combination of everolimus and bevacizumab was shown to be well tolerated and had a 26 % response rate in patients with advanced NET [73]. Given these results, the National Cancer Institute (NCI) has an ongoing phase II study randomizing patients with locally advanced or metastatic pNETs not amenable to surgery to receive everolimus and octreotide with or without bevacizumab to assess antitumor activity and toxicity of the regimen (CALGB 80701; NCT01229943).
The PI3K oncogene plays an essential role in the PI3K/Akt/mTOR signaling pathway. Three classes of PI3K have been described. Class I enzymes are activated directly by cell-surface receptors, and the catalytic domain of class IA PI3K p110 subunits are widely implicated in cancer [74]. The constitutive activation of the mTOR pathway from mutation and overexpression of PI3K or one of its components can potentially lead to tumorigenesis [75]. Dual inhibitors of both PI3K and mTOR are emerging as the catalytic domain of mTOR is structurally similar to catalytic domains of the PI3K p110 subunits. Unlike the rapalogs, these dual inhibitors suppress both the mTORC1 and mTORC2 complex.
A new generation of mTOR inhibitors is being developed which may bypass feedback loops and address mTORC2-mediated escape mechanisms and resistance, potentially increasing their efficacy compared with rapalogs. The dual mTORC1/2 and PI3K inhibitor NVP-BEZ235 (Novartis, East Hanover, NJ) has demonstrated antiproliferative activity against a variety of cancer and has shown to be a more efficient inducer of apoptosis and cell cycle arrest than single inhibitors in various NET cell lines. After treatment with everolimus, NVP-BEZ235 prevented both vertical and horizontal negative feedback activation of Akt [76, 77]. Data from phase I clinical trial of NVP-BEZ235 did not show any dose-limiting toxicity in the first 59 treated patients. There is currently an ongoing phase II study evaluating NVP-BEZ235 plus best supportive care versus placebo plus best supportive care in patients with advanced pNET after failure of mTOR inhibitor therapy (NCT01658436).
The combination of everolimus and the RAF inhibitor RAF265 was also more effective than treatment with a single kinase inhibitor. RAF265 not only inhibited ERK1/2 phosphorylation, but also strongly induced Akt phosphorylation and VEGF secretion due to Akt-mediated HIF-1α activation. NVP-AEW541 is a novel selective IGF-1R tyrosine kinase inhibitor that inhibits the key upstream receptor for IGF-1 signaling to target both the PI3K/Akt/mTOR and RAS/RAF/MEK pathways [46]. It has been shown to be active in BON cells and a human insulinoma cell line. The antineoplastic effects of NVP-AEW541 involve the inactivation of ERK1/2. NVP-AEW541 caused apoptosis and cell cycle arrest and inhibited NET cell proliferation in a dose-dependent fashion. Moreover, there was an increase in the antiproliferative properties when NVP-AEW541 was combined with doxorubicin and fluvastatin [78].
5.6 Predictive Biomarkers
Surrogate markers to assess response to targeted therapy are needed since traditional staging modalities may not reflect actual response to therapy. There are currently no definitive PI3K/Akt/mTOR pathway biomarkers that predict response to mTOR inhibitors. Evaluating multiple markers may help identify oncogenic signaling drivers of each tumor. Preclinical assays have identified S6K1 as a molecular marker that is currently assessed in clinical trials [79]. For example, temsirolimus was shown to reduce the phosphorylation of S6K1 (p-S6K1) both in peripheral blood mononuclear cells and tumor biopsies [30]. Inhibition of mTOR signaling detected as a reduction of p-S6K1 (−92.5 to +100 % of initial values) and p-4EBP1 (−5.9 to −63.8 % of initial values) was also seen in tumor biopsies performed after administration of everolimus. There was also a significant reduction of p-S6K1 in peripheral blood cells [45, 80]. S6K1 and 4EBP1 phosphorylation can be used as a surrogate marker to assess efficacy of mTOR inhibitors in skin, blood, and tumor samples.
Other biomarkers for mTOR pathway inhibitors have also been investigated in neuroendocrine tumors. IGFR-1 overexpression has been linked to upregulation of the mTOR pathway. Casanovas et al. described the activation of this pathway by immunohistochemistry in 69 tumor samples of NETs [81]. IGF1R was expressed in 66 % and phosphorylated mTOR (p-mTOR) was only expressed in 20 %. A subgroup of midgut NETs showed consistent activation of both IGF1R and p-mTOR. Another study by Heetfield et al. analyzed 26 cases of GEP-NETs for p-mTOR and phosphorylated eukaryotic translation initiation factor 4E (p-eIF4E) [82]. p-mTOR was expressed in 64 % and its downstream effector p-eIF4E was expressed in 24 %. High expression levels of these biomarkers were significantly associated with shorter survival. IGFR-1 expression, p-mTOR, and p-eIF4E may be relevant biomarkers for the selection of inhibitors of the mTOR pathway, and preliminary data suggest the need for further research.
Recently, a single nucleotide polymorphism (snp) in the fibroblast growth factor receptor (4 FGFR4-G388R) was reported to be prognostic and predictive for pancreatic neuroendocrine tumors [83]. This snp was identified in 36/71 patients and correlated with poor survival and deceased efficacy for treatment with the mTOR inhibitor everolimus. This association was confirmed with preclinical models of transfected pancreatic neuroendocrine cancer cell lines.
5.7 Conclusion
In summary, NETs are diverse and heterogeneous in underlying tumor biology and clinical presentations. Before the development of targeted agents, limited options were available to control tumor growth and improve patient’s quality of life. Due to our better understanding of the various molecular signaling pathways involved in NET growth, there are now several emerging treatment options. For the first time in 20 years, new agents have been approved for treatment of pNETs that target the PI3K/Akt/mTOR pathway.
The PI3K/Akt/mTOR pathway plays a critical role in regulating cell growth and apoptosis. mTOR is a novel and validated molecular target in the treatment of NETs. The success of everolimus in prolonging PFS in pNET supports targeting the PI3K/Akt/mTOR pathway as an important strategy for making therapeutic advances in NETs. There are currently several ongoing clinical trials exploring the role of second-generation mTOR inhibitors as well as rationale combination regimens. Biomarkers to enhance the efficacy of these drugs are being actively pursued and are making their way into practice. These efforts will ultimately change the way we treat NETs and other malignant tumors.
References
Jensen RT, Delle Fave G. Promising advances in the treatment of malignant pancreatic endocrine tumors. N Engl J Med. 2011;364(6):564–5.
Metz DC, Jensen RT. Gastrointestinal neuroendocrine tumors: pancreatic endocrine tumors. Gastroenterology. 2008;135(5):1469–92.
Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063–72.
Tsikitis VL, Wertheim BC, Guerrero MA. Trends of incidence and survival of gastrointestinal neuroendocrine tumors in the United States: a seer analysis. J Cancer. 2012;3:292–302.
Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934–59.
Jensen RT, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer. 2008;113(7 Suppl):1807–43.
Kaltsas GA, Besser GM, Grossman AB. The diagnosis and medical management of advanced neuroendocrine tumors. Endocr Rev. 2004;25(3):458–511.
Oberg K, Kvols L, Caplin M, Delle Fave G, de Herder W, Rindi G, et al. Consensus report on the use of somatostatin analogs for the management of neuroendocrine tumors of the gastroenteropancreatic system. Ann Oncol. 2004;15(6):966–73.
Rinke A, Müller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656–63.
Wolin EM. PI3K/Akt/mTOR pathway inhibitors in the therapy of pancreatic neuroendocrine tumors. Cancer Lett. 2013;335(1):1–8.
Shida T, Kishimoto T, Furuya M, Nikaido T, Koda K, Takano S, et al. Expression of an activated mammalian target of rapamycin (mTOR) in gastroenteropancreatic neuroendocrine tumors. Cancer Chemother Pharmacol. 2010;65(5):889–93.
Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331(6021):1199–203.
Ghayouri M, Boulware D, Nasir A, Strosberg J, Kvols L, Coppola D. Activation of the serine/theronine protein kinase Akt in enteropancreatic neuroendocrine tumors. Anticancer Res. 2010;30(12):5063–7.
Missiaglia E, Dalai I, Barbi S, Beghelli S, Falconi M, della Peruta M, et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28(2):245–55.
Yao JC. Neuroendocrine tumors. Molecular targeted therapy for carcinoid and islet-cell carcinoma. Best Pract Res Clin Endocrinol Metab. 2007;21(1):163–72.
Fazio N, Cinieri S, Lorizzo K, Squadroni M, Orlando L, Spada F, et al. Biological targeted therapies in patients with advanced enteropancreatic neuroendocrine carcinomas. Cancer Treat Rev. 2010;36 Suppl 3:S87–94.
Carew JS, Kelly KR, Nawrocki ST. Mechanisms of mTOR inhibitor resistance in cancer therapy. Target Oncol. 2011;6(1):17–27.
Durán I, Salazar R, Casanovas O, Arrazubi V, Vilar E, Siu LL, et al. New drug development in digestive neuroendocrine tumors. Ann Oncol. 2007;18(8):1307–13.
Zhang J, Jia Z, Li Q, Wang L, Rashid A, Zhu Z, et al. Elevated expression of vascular endothelial growth factor correlates with increased angiogenesis and decreased progression-free survival among patients with low-grade neuroendocrine tumors. Cancer. 2007;109(8):1478–86.
Meric-Bernstam F, Akcakanat A, Chen H, Do KA, Sangai T, Adkins F, et al. PIK3CA/PTEN mutations and Akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clin Cancer Res. 2012;18(6):1777–89.
Shah T, Hochhauser D, Frow R, Quaglia A, Dhillon AP, Caplin ME. Epidermal growth factor receptor expression and activation in neuroendocrine tumours. J Neuroendocrinol. 2006;18(5):355–60.
Wang L, Ignat A, Axiotis CA. Differential expression of the PTEN tumor suppressor protein in fetal and adult neuroendocrine tissues and tumors: progressive loss of PTEN expression in poorly differentiated neuroendocrine neoplasms. Appl Immunohistochem Mol Morphol. 2002;10(2):139–46.
Pitt SC, Davis R, Kunnimalaiyaan M, Chen H. AKT and PTEN expression in human gastrointestinal carcinoid tumors. Am J Transl Res. 2009;1(3):291–9.
Wang Y, Ozawa A, Zaman S, Prasad NB, Chandrasekharappa SC, Agarwal SK, et al. The tumor suppressor protein menin inhibits AKT activation by regulating its cellular localization. Cancer Res. 2011;71(2):371–82.
Corbo V, Beghelli S, Bersani S, Antonello D, Talamini G, Brunelli M, et al. Pancreatic endocrine tumours: mutational and immunohistochemical survey of protein kinases reveals alterations in targetable kinases in cancer cell lines and rare primaries. Ann Oncol. 2012;23(1):127–34.
Corbo V, Dalai I, Scardoni M, Barbi S, Beghelli S, Bersani S, et al. MEN1 in pancreatic endocrine tumors: analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr Relat Cancer. 2010;17(3):771–83.
Toliat MR, Berger W, Ropers HH, Neuhaus P, Wiedenmann B. Mutations in the MEN I gene in sporadic neuroendocrine tumours of gastroenteropancreatic system. Lancet. 1997;350(9086):1223.
Fujii T, Kawai T, Saito K, Hishima T, Hayashi Y, Imura J, et al. MEN1 gene mutations in sporadic neuroendocrine tumors of foregut derivation. Pathol Int. 1999;49(11):968–73.
Kasajima A, Pavel M, Darb-Esfahani S, Noske A, Stenzinger A, Sasano H, et al. mTOR expression and activity patterns in gastroenteropancreatic neuroendocrine tumours. Endocr Relat Cancer. 2011;18(1):181–92.
Duran I, Kortmansky J, Singh D, Hirte H, Kocha W, Goss G, et al. A phase II clinical and pharmacodynamic study of temsirolimus in advanced neuroendocrine carcinomas. Br J Cancer. 2006;95(9):1148–54.
Fjällskog ML, Lejonklou MH, Oberg KE, Eriksson BK, Janson ET. Expression of molecular targets for tyrosine kinase receptor antagonists in malignant endocrine pancreatic tumors. Clin Cancer Res. 2003;9(4):1469–73.
Zhang L, Smyrk TC, Oliveira AM, Lohse CM, Zhang S, Johnson MR, et al. KIT is an independent prognostic marker for pancreatic endocrine tumors: a finding derived from analysis of islet cell differentiation markers. Am J Surg Pathol. 2009;33(10):1562–9.
Nilsson O, Wängberg B, Theodorsson E, Skottner A, Ahlman H. Presence of IGF-I in human midgut carcinoid tumours–an autocrine regulator of carcinoid tumour growth? Int J Cancer. 1992;51(2):195–203.
von Wichert G, Jehle PM, Hoeflich A, Koschnick S, Dralle H, Wolf E, et al. Insulin-like growth factor-I is an autocrine regulator of chromogranin A secretion and growth in human neuroendocrine tumor cells. Cancer Res. 2000;60(16):4573–81.
Wang Y, Sun Y. Insulin-like growth factor receptor-1 as an anti-cancer target: blocking transformation and inducing apoptosis. Curr Cancer Drug Targets. 2002;2(3):191–207.
Lamberts SW, van der Lely AJ, de Herder WW, Hofland LJ. Octreotide. N Engl J Med. 1996;334(4):246–54.
Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648–57.
Capdevila J, Tabernero J. A shining light in the darkness for the treatment of pancreatic neuroendocrine tumors. Cancer Discov. 2011;1(3):213–21.
Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5(8):671–88.
Bukowski RM. Temsirolimus: a safety and efficacy review. Expert Opin Drug Saf. 2012;11(5):861–79.
Hobday TJ, Qin R, Moore MJ, Reidy DL, Strosbery JR, Kindler HL, et al. Multicenter phase II trial of temsirolimus (TEM) and bevacizumab (BEV) in pancreatic neuroendocrine tumor (PNET). J Clin Oncol. 2013;31(Suppl; abstr 4032).
Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514–23.
Zitzmann K, De Toni EN, Brand S, Göke B, Meinecke J, Spöttl G, et al. The novel mTOR inhibitor RAD001 (everolimus) induces antiproliferative effects in human pancreatic neuroendocrine tumor cells. Neuroendocrinology. 2007;85(1):54–60.
Grozinsky-Glasberg S, Franchi G, Teng M, Leontiou CA, Ribeiro de Oliveira A, Dalino P, et al. Octreotide and the mTOR inhibitor RAD001 (everolimus) block proliferation and interact with the Akt-mTOR-p70S6K pathway in a neuro-endocrine tumour cell Line. Neuroendocrinology. 2008;87(3):168–81.
Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol. 2008;26(10):1603–10.
Yao JC, Phan AT, Chang DZ, Wolff RA, Hess K, Gupta S, et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: results of a phase II study. J Clin Oncol. 2008;26(26):4311–8.
Yao JC, Lombard-Bohas C, Baudin E, Kvols LK, Rougier P, Ruszniewski P, et al. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: a phase II trial. J Clin Oncol. 2010;28(1):69–76.
Pavel ME, Hainsworth JD, Baudin E, Peeters M, Hörsch D, Winkler RE, et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. Lancet. 2011;378(9808):2005–12.
Ito T, Okusaka T, Ikeda M, Igarashi H, Morizane C, Nakachi K, et al. Everolimus for advanced pancreatic neuroendocrine tumours: a subgroup analysis evaluating Japanese patients in the RADIANT-3 trial. Jpn J Clin Oncol. 2012;42(10):903–11.
Pavel M, Baudin E, Couvelard A, Krenning E, Öberg K, Steinmüller T, et al. ENETS Consensus Guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology. 2012;95(2):157–76.
Svejda B, Kidd M, Kazberouk A, Lawrence B, Pfragner R, Modlin IM. Limitations in small intestinal neuroendocrine tumor therapy by mTor kinase inhibition reflect growth factor-mediated PI3K feedback loop activation via ERK1/2 and AKT. Cancer. 2011;117(18):4141–54.
Yao P, Fogleman N, Jacobs D, et al. Randomized run-in study of bevacizumab (B) and everolimus (E) in low- to intermediate-grade neuroendocrine tumors (LGNETs) using perfusion CT as functional biomarker. Chicago: American Society of Clinical Oncology; 2010.
Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27(41):5527–41.
Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166(2):213–23.
Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway–beyond rapalogs. Oncotarget. 2010;1(7):530–43.
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–44.
Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125(4):733–47.
O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500–8.
Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26(13):1932–40.
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–19.
Mita M, Wolin E, Meyer T, Nemunaitis J, Bergsland E, Mahipal A, et al. Phase I expansion trial of an oral TORC1/TORC2 inhibitor (CC-223) in nonpancreatic neuroendocrine tumors (NET). J Clin Oncol. 2013;31(Suppl; abstr e15004).
Gloesenkamp C, Nitzsche B, Lim AR, Normant E, Vosburgh E, Schrader M, et al. Heat shock protein 90 is a promising target for effective growth inhibition of gastrointestinal neuroendocrine tumors. Int J Oncol. 2012;40(5):1659–67.
Tolcher AW, Sarantopoulos J, Patnaik A, Papadopoulos K, Lin CC, Rodon J, et al. Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. J Clin Oncol. 2009;27(34):5800–7.
Beltran PJ, Mitchell P, Chung YA, Cajulis E, Lu J, Belmontes B, et al. AMG 479, a fully human anti-insulin-like growth factor receptor type I monoclonal antibody, inhibits the growth and survival of pancreatic carcinoma cells. Mol Cancer Ther. 2009;8(5):1095–105.
Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9(7):1956–67.
Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29(35):4688–95.
Gloesenkamp CR, Nitzsche B, Ocker M, Di Fazio P, Quint K, Hoffmann B, et al. AKT inhibition by triciribine alone or as combination therapy for growth control of gastroenteropancreatic neuroendocrine tumors. Int J Oncol. 2012;40(3):876–88.
Zitzmann K, Vlotides G, Brand S, Lahm H, Spöttl G, Göke B, et al. Perifosine-mediated Akt inhibition in neuroendocrine tumor cells: role of specific Akt isoforms. Endocr Relat Cancer. 2012;19(3):423–34.
Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27(13):2278–87.
Yuan R, Kay A, Berg WJ, Lebwohl D. Targeting tumorigenesis: development and use of mTOR inhibitors in cancer therapy. J Hematol Oncol. 2009;2:45.
Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol. 2005;16(4):525–37.
Villaume K, Blanc M, Gouysse G, Walter T, Couderc C, Nejjari M, et al. VEGF secretion by neuroendocrine tumor cells is inhibited by octreotide and by inhibitors of the PI3K/AKT/mTOR pathway. Neuroendocrinology. 2010;91(3):268–78.
Kulke MH, Siu LL, Tepper JE, Fisher G, Jaffe D, Haller DG, et al. Future directions in the treatment of neuroendocrine tumors: consensus report of the National Cancer Institute Neuroendocrine Tumor clinical trials planning meeting. J Clin Oncol. 2011;29(7):934–43.
Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. 1999;253(1):239–54.
Jimenez C, Jones DR, Rodríguez-Viciana P, Gonzalez-García A, Leonardo E, Wennström S, et al. Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3-kinase. EMBO J. 1998;17(3):743–53.
Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68(22):9221–30.
Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68(19):8022–30.
Reidy-Lagunes DL, Vakiani E, Segal MF, Hollywood EM, Tang LH, Solit DB, et al. A phase 2 study of the insulin-like growth factor-1 receptor inhibitor MK-0646 in patients with metastatic, well-differentiated neuroendocrine tumors. Cancer. 2012;118(19):4795–800.
Boulay A, Zumstein-Mecker S, Stephan C, Beuvink I, Zilbermann F, Haller R, et al. Antitumor efficacy of intermittent treatment schedules with the rapamycin derivative RAD001 correlates with prolonged inactivation of ribosomal protein S6 kinase 1 in peripheral blood mononuclear cells. Cancer Res. 2004;64(1):252–61.
Raymond E, Alexandre J, Faivre S, Vera K, Materman E, Boni J, et al. Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI-779, a novel mTOR inhibitor, in patients with cancer. J Clin Oncol. 2004;22(12):2336–47.
Casanovas O, Teule A, Villabona C. Study on activation of the IGF-1R mTOR pathway in neuroendocrine tumours (NETs). ASCO annual conference 2013. Chicago: Journal of Clinical Oncology; 2013.
Heetfeld M, Kasajima A, Koch A. Poorly differentiated neuroendocrine carcinoma (NEC G3): prognostic factors and potential novel targets. American Society of Clinical Oncology annual conference. Chicago, 2013. p. #e15071.
Serra S, Zheng L, Hassan M, Phan AT, Woodhouse LJ, Yao JC, et al. The FGFR4-G388R single-nucleotide polymorphism alters pancreatic neuroendocrine tumor progression and response to mTOR inhibition therapy. Cancer Res. 2012;72(22):5683–91.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer-Verlag France
About this chapter
Cite this chapter
Hendifar, A.E., Liu, S., Wolin, E. (2016). The Role of mTOR Inhibitors in Neuroendocrine Tumors. In: Mita, M., Mita, A., Rowinsky, E. (eds) mTOR Inhibition for Cancer Therapy: Past, Present and Future. Springer, Paris. https://doi.org/10.1007/978-2-8178-0492-7_5
Download citation
DOI: https://doi.org/10.1007/978-2-8178-0492-7_5
Publisher Name: Springer, Paris
Print ISBN: 978-2-8178-0491-0
Online ISBN: 978-2-8178-0492-7
eBook Packages: MedicineMedicine (R0)