
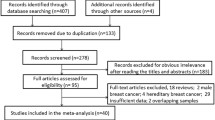
Abstract
Promoter-CpG island hypermethylation has been proposed as an alternative mechanism to inactivate BRCA1 in the breast where somatic mutations of BRCA1 are rare. To better understand breast cancer etiology and progression, we explored the association between BRCA1 promoter methylation status and prognostic factors as well as survival among women with breast cancer. Promoter methylation of BRCA1 was assessed in 851 archived tumor tissues collected from a population-based study of women diagnosed with invasive or in situ breast cancer in 1996–1997, and who were followed for vital status through the end of 2002. About 59% of the tumors were methylated at the promoter of BRCA1. The BRCA1 promoter methylation was more frequent in invasive cancers (P = 0.02) and among premenopausal cases (P = 0.05). BRCA1 promoter methylation was associated with increased risk of breast cancer-specific mortality (age-adjusted HR 1.71; 95% CI: 1.05–2.78) and all-cause mortality (age-adjusted HR 1.49; 95% CI: 1.02–2.18). Neither dietary methyl intakes in the year prior to the baseline interview nor the functional polymorphisms in one-carbon metabolism were associated with BRCA1 methylation status. Our study is the first epidemiological investigation on the prognostic value of BRCA1 promoter methylation in a large population-based cohort of breast cancer patients. Our results indicate that BRCA1 promoter methylation is an important factor to consider in predicting breast cancer survival.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the leading cause of cancer mortality among women 20–59 years of age and the second leading cause of cancer mortality among all women [1]. Breast cancer is a manifestation of abnormal genetic as well as epigenetic changes [2]. Promoter-CpG island hypermethylation, accompanied by global hypomethylation, are common molecular defects in cancer cells [3, 4]. Although the causal relationship is still being debated, evidence has shown that hypermethylation is associated with silencing of many crucial genes in the neoplastic process [5]. This phenomenon has also been reported in a large panel of genes in breast cancer [6].
Breast cancer gene 1(BRCA1), located on chromosome 17q21 (Fig. 1), encodes a multifunctional protein involved in DNA repair, control of cell-cycle checkpoints, protein ubiquitinylation and chromatin remodeling [7]. It was originally cloned as a gene responsible for familial breast cancer [8]. About 5–50% of familial breast cancers could be explained by inherited mutations of BRCA1 in different populations [9]. However, somatic mutations of BRCA1 are rare in sporadic breast cancers despite the high degree of loss of heretozygosity (LOH) at this locus [6, 10]. Therefore, other mechanisms for loss of function must exist. DNA methylation has been proposed as an alternative mechanism to inactivate BRCA1 [11]. Results from various methods of detection revealed that 9–44% of breast cancer samples harbored a hypermethylated promoter at BRCA1 [11, 12].

Schematic illustration of the BRCA1 promoter region for methylation analysis. The view of the genomic context was adapted from USCS Genome Browsers (http://genome.uscs.edu). The reverse strand of BRCA1 is shown. The blue bar shows the exons of the RefSeq Genes and the green bar shows the predicted CpG island in the promoter region. Location of transcription start sites (TSS) is also shown. An enlarged view of the region examined (Exon 1 and nearby region) shows the location of the primers for methylation-specific PCR and CpG sites (red vertical bars)
BRCA1 status may potentially be used as a prognostic marker as studies have shown that breast cancers with BRCA1 mutations are usually poorly differentiated, highly proliferative, ER-, PR-, and harbor p53 mutations [13]. BRCA1 mutated breast cancer is also associated with poor survival in some studies [14–18].
One-carbon metabolism may be involved in the DNA methylation process as it provides the universal methyl donor, S-adenosylmethionine (SAM). Folate, methionine and choline are the major sources of methyl groups in foods [19]. There is evidence that dietary methyl donors are capable of modulating methylation patterns in both animal models and humans [20–23]. Furthermore, functional polymorphisms in one-carbon metabolizing genes could in principle modify DNA methylation status [24–26].
We previously reported that intakes of B vitamins as well as common polymorphisms in one-carbon metabolizing genes were associated with breast cancer risk in the population-based Long Island Breast Cancer Study Project [27–29]. Herein, we investigated promoter methylation status of BRCA1 in relation to clinical/pathological factors and breast cancer survival in the same population. The influence of dietary methyl intake as well as polymorphisms in one-carbon metabolizing genes on BRCA1 promoter methylation was also examined.
Materials and methods
Study population
We utilized the resources of the parent case-control as well as the follow-up study of the Long Island Breast Cancer Study Project, a population-based study. The study participants included women newly diagnosed with a first primary breast cancer who participated in the original case-control study [30] and were subsequently re-interviewed about five years later and followed for vital status [31]. Details of the study design have been described in detail previously [30–33].
Exposure data was obtained as part of the (1) case-control (baseline) interview; (2) follow-up interview; and (3) medical record abstraction. The questionnaires were administrated to assess the demographic characteristics, breast cancer-related factors, tumor characteristics and treatment information. The study protocol was approved by the Institutional Review Boards of the collaborating institutions. REMARK criteria were used through this report [34].
Study outcome
The National Death Index was used to ascertain all-cause and breast cancer-specific mortality. Among the 1508 women diagnosed with breast cancer in 1996–1997, 198 (13.1%) deaths occurred by December 31, 2002. The mean follow up time was 5.6 years (range: 0.2–7.4). Based on International Classification of Diseases (ICD) codes 174.9 and C–50.9 listed as a primary or secondary code on the death certificate, 124 (62.6%) of these 198 deaths were due to breast cancer.
Tumor block retrieval and DNA extraction
Tumor tissue blocks were requested from all 35 participating hospitals of the parent study. In total, breast cancer tissue blocks were successfully retrieved for 975 case participants (67.2%). We compared the demographic and clinicopathological features between cases with or without tumor block available for methylation analysis in our study. Although most characteristics are similar between these two groups, some factors were different. Case women who had tumor samples available for methylation analysis tended to be older (mean age 59.6 vs. 57.9; P = 0.005); to have an invasive tumor (87.8% vs. 80.1%; P < 0.001); and to be post-menopausal (70.7% vs. 64.6%; P = 0.01).
The paraffin blocks from each case participant were used to generate 15x 5 micron and 10x 10 micron thick slides. Tumor tissues were isolated from 10 micron paraffin sections by microdissection. Tumor DNA was isolated by adding 30 ul of proteinase K-digestion buffer (50 mM Tris, pH 8.1, 1 mM EDTA, 0.5% Tween 20, 10 μg/ml proteinase K) to the tube and by incubating overnight at 37°C. Proteinase K was inactivated incubating the samples at 95°C for 10 min and centrifugation.
Analysis of BRCA1 promoter methylation
BRCA1 promoter methylation was determined by methylation-specific PCR (MSP) with bisulfite-converted DNA (illustrated in Fig. 1). Bisulfite modification of DNA to convert unmethylated cytosine residues to uracil was carried out using the CpGnome DNA Modification Kit (Chemicon International, Purchase, NY) following the protocol from the manufacturer. Sequences of the primers were: (i) Methylated primer: forward-5′-GAG AGG TTG TTG TTT AGC GG-3′; backward-5′-CGC GCA ATC GCA ATT TTA AT-3′; (ii) Unmethylated primers: forward-5′-TGG TAA TGG AAA AGT GTG GGA A-3′; backward-5′-CCC ATC CAA AAA ATC TCA ACA AA-3′. PCR was carried out in a total volume of 20 μl containing 0.5 U of AmpliTaq Gold II (Roche, Nutley, NJ, USA). The amplicon was 146 bp in length. Each PCR reaction underwent initial denaturation at 95°C for 10 min, and 40 cycles of the following profile: 30 s at 94°C, 30 s at 55°C, and 30 s at 72°C followed by a final 10 min extension at 72°C. The PCR products were then analyzed by electrophoresis on a 2% agarose gel, stained with ethidium bromide and visualized by UV transillumination. DNA is considered methylated if a PCR product using methylated-specific primers was visualized while a PCR product using unmethylated-specific primers is absent. Bisulfite-modified universal methylated DNA (Chemicon International, NY) and sperm DNA were used as methylated and unmethylated controls, respectively. The assay was successfully completed for 851 subjects; the main reason for failure of methylation assessment was insufficient DNA from tumor blocks.
Dietary assessment
As part of the baseline interview, participants were asked to complete a modified Block food frequency questionnaire (FFQ), which assessed intake of over 100 food items in the year prior to the interview [33]. The frequency and portion size data were translated to daily intakes of nutrients from both dietary and supplement sources using the National Cancer Institute’s DietSys version 3 for folate, and a previously described protocol for choline, methionine and betaine [35]. Habitual use of multivitamin supplements was also obtained from the FFQ. Dietary intake values for one-carbon related micronutrients and compounds were calculated based on interview data assessed from this FFQ.
Blood sample collection and genotyping
Blood samples were collected from 73% of the cases at the time of the baseline interview by trained field staff [30] and DNA was isolated from blood specimens using the methods previously described [32]. Genotyping was conducted on 9 polymorphisms in the one-carbon metabolism pathway using methods described elsewhere [27, 28].
Statistical analysis
Correlation of BRCA1 promoter methylation status of the tumor tissue with patient demographic characteristics, other factors that may affect prognosis, and with known characteristics of the breast cancer diagnosis was examined using the chi-square statistic for categorical variables and by two-sample t-test for continuous variables. The Kaplan-Meier and the log-rank test were used to examine the crude association between BRCA1 promoter methylation status and survival [36]. The Cox proportional hazard regression [36] was used to estimate the hazard ratio (HR) and 95% confidence interval (CI) for breast cancer-specific and all-cause mortality, with adjustments made for age at diagnosis (continuous). Potential confounding effect by other factors known to influence survival among breast cancer patients was evaluated by adjustment in the Cox model. These factors include age at diagnosis, cancer type (in situ vs. invasive), menopausal status (pre- vs. post-), race, family history of breast cancer and history of benign breast. One-carbon metabolism-related nutrient intakes in the year prior to the interview were divided into quintiles based on the distributions observed in cases with methylation data. Since we anticipate that only those with extreme low intake may have the phenotypes of interest, we compared the incidence of higher methylation in the very low nutrient intake group, with that the pool of the other four qunitiles. Nutrients examined in the study include folate, methionine, choline, betaine and B vitamins (B1, B2, B3, B6, B12). Whether the distribution of the one-carbon genotypes differed with respect to BRCA1 methylation status was examined using the chi-square statistic. All statistical analyses were performed using SAS statistical software version 9.1(SAS Institute, Cary, NC).
Results
BRCA1 methylation and clinicopathological characteristics of breast cancer
Promoter methylation status of BRCA1 was assessed in breast tumor samples from a population-based sample of 851 women with breast cancer, including 104 in situ and 747 invasive cases (Table 1). Overall 504/851 (59.2%) of tumors showed methylation at the promoter of BRCA1. Table 1 summarizes the relationship between BRCA1 methylation status and clinicopathological, lifestyle factors. BRCA1 promoter methylation was more frequent in cancers that were classified as invasive (P = 0.02) and among premenopausal women (P = 0.05). BRCA1 promoter methylation was not associated with age at diagnosis or family history of breast cancer.
Hormone receptor status, as recorded on the medical record, was determined by immunohistochemistry; this information was available on 625 out of 851 samples (Table 2). BRCA1 promoter methylation status was not associated with ER or PR status in this population. For a smaller subset of women, we were able to obtain information on tumor size (n = 321) and node involvement (n = 327) from the medical record. BRCA1 promoter methylation was more frequent in cancers with at least one node involved (P = 0.003) and with tumor size greater than 2 cm (P = 0.003).
BRCA1 methylation and survival
Among the 851 women with methylation data available, a total of 122 (14.3%) deaths were observed; 79 (64.8%) of these were due to breast cancer. As shown in Fig. 2, BRCA1 methylation was associated with breast cancer-specific mortality among the cohort of women in our analysis (P for log-rank test = 0.03). Compared to cases with an unmethylated BRCA1 promoter, those who had a methylated BRCA1 promoter had a 72% increased risk of dying from breast cancer at the end of follow up (age-adjusted HR: 1.72, 95% CI: 1.06–2.79). A similar result was observed for all-cause mortality with borderline significance (p for log-rank test = 0.05); cases with methylated BRCA1 promoters had a 45% increased mortality risk when compared to those who had unmethylated BRCA1 promoters (age-adjusted HR: 1.45, 95% CI: 0.99–2.11). The BRCA1 methylation and survival association was of borderline significance after adjusting for age, cancer type (in situ vs. invasive), menopausal status (pre- vs. post-), race, family history of breast cancer and history of benign breast disease in a multivariate model (multi-variate adjusted HR for breast cancer-specific mortality: 1.67, 95% CI: 0.99–2.81; for all-cause mortality: 1.40, 95% CI: 0.94–2.08).

Survival plot for breast cancer patients by BRCA1 promoter methylation status in the tumor tissue—Kaplan–Meier analyses of survival among all 851 breast cancer cases. Vertical lines in the curve represent the death events
Among cases that have BRCA1 methylation status assessed, information on whether they received chemotherapy, radiation therapy or hormore therapy was available on ~550 cases. When survival analysis was performed according to treatment groups, the relationship between BRCA1 methylation and survival association dose not differ with treatment.
One-carbon metabolism and BRCA1 methylation
We explored whether dietary intakes of one-carbon related nutrients a year prior to baseline interview were associated with BRCA1 methylation status. None of the nutrient intakes was associated with BRCA1 methylation. Furthermore, we did not observe any relationships between BRCA1 promoter methylation and functional polymorphisms in the one-carbon metabolizing genes [(MTHFR C677T (rs1801133) and A1298C (rs1801131); TYMS 5’-UTR tandem repeat; DHFR 19 bp deletion; MTR A2756 (rs1805087); MTRR A66G (rs1801394); BHMT G742A (rs3733890); RFC1 A80G (rs1051266); and cSHMT C1420T (rs1979277)] (data not shown).
Discussion
To reduce the disease burden of breast cancer, it is important to identify etiologic factors of the disease as well as factors that influence survival. We studied BRCA1 promoter methylation because the importance of this gene has been well documented in breast carcinogenesis [37]. To the best of our knowledge, this is the first epidemiologic study on the prognostic value of BRCA1 methylation. The cohort of women with breast cancer was drawn from a large population-based sample that encompassed a broad age range making the study results more generalizable than a series of cases from a single institution. Furthermore, the comprehensive lifestyle and dietary information as well as collection of biological samples allowed us to examine interactions among environmental, genetic, and epigenetic factors in relation to breast cancer mortality.
We found BRCA1 promoter methylation in ~59% of tumors, a level higher than published studies from other groups, which ranged from 9–44% [11, 12]. Several factors may account for these differences. First, the assay used for methylation measurement varied from study to study. Because contamination from adjacent tissue may occur during tissue dissection, unmethylated DNA from the normal cells might attenuate the methylation levels of the tumor tissue. In our study, tumor tissues were micro-dissected from paraffin-embedded sections so the contamination was minimized. Secondly, MSP detects differential methylation status by amplification of bisulfite-treated DNA with primers specific for methylated vs. unmethylated DNA [38]. CpG sites residing within the primer sets were used as a proxy for the methylation status of the region of interest. Although most published studies mentioned above used MSP, the primer sequences and target regions varied from study to study. Considering the specificity of our assay, we demonstrated that by incorporating both methylated and unmethylated DNA as controls.
We found that BRCA1 promoter methylation was more frequent in invasive than in in situ carcinomas. Since information on breast cancer subtype was not readily available for the other published studies, we could not compare the methylation-cancer type relationship observed here directly with other studies. In a small subset of our population for whom medical record data were available, we also found a higher prevalence of BRCA1 promoter methylation in cases with at least one node involved and with tumor size greater than 2 cm. Taken together, these associations suggest that methylation occurs in sequence during tumor development and progression. Higher methylation levels could result in a more advanced tumor stage at diagnosis. Thus, it is possible that the variation in methylation prevalence reported across studies could also be due to the variation in the distributions of cases’ stage at diagnosis.
We found that BRCA1 promoter methylation was more frequent in tumors from premenopausal women despite the fact that age of diagnosis was not associated with BRCA1 methylation. Studies have indicated that estrogens stimulate the expression of BRCA1 [39]. On the other hand, BRCA1 was shown to have an ability to inhibit the cellular response to estrogens by direct interaction with the estrogen receptor [40, 41]. Nevertheless, we did not observe any association between BRCA1 methylation and ER/PR status in our study.
BRCA1 mutation status as a potential prognostic marker had been explored previously. A recent review by Liebens et al. [42] summarized differences in survival outcome in relation to BRCA1 germline mutations. Evidence exists indicating that BRCA1 mutation carriers had a worse survival [14–18]. Carriers of BRCA1 mutations had functionally defective proteins, resulting in some loss of function. Promoter methylation represented an alternative mechanism for loss of function of BRCA1. Our study is the first to report that BRCA1 promoter methylation influences breast cancer survival in an epidemiologic study. Decreased survival associated with methylated BRCA1 corroborates the findings on BRCA1 mutations. This finding may have clinical significance as it identifies a subgroup of patients with worse survival and could help in tailoring of breast cancer treatment based on epigenetic profiles.
We found no association between intake of nutrients involved with one-carbon metabolism and BRCA1 methylation status. Although previous studies have shown that dietary methyl content and one-carbon gene polymorphisms were capable of modulating global DNA methyl content in animal models and humans [20–26], modulation of promoter methylation of specific genes has not been demonstrated. Since epigenetic changes may be reversible and diet is a modifiable factor, further investigation into this relationship could aid in our search for breast cancer chemoprevention strategies.
One limitation of our study is that BRCA1 expression was not measured in the tumor tissues. Thus, we could not explore the functional consequence of DNA methylation. Previous studies have generated conflicting results on BRCA1 expression and the subcellular localization of this protein by using various antibodies for immunohistochemical staining [43–45]. This discrepancy was even more pronounced when paraffin-embedded tissues were used [46]. Nevertheless, the fact that our results corroborate those from BRCA1 mutation studies suggests that promoter methylation indeed results in gene silencing or loss of gene function.
Another limitation of our study is that tumor DNA was not available for all case participants of the Long Island Breast Cancer Study Project, which is a population-based study. Although there were some differences between those with and without tumor DNA available for our analyses (described in the Methods section), the benefit of utilizing our population-based sample is that we are able to quantify the differences between the two groups. This valuable contrast aids in our interpretation of the generalizability of our study results to the general population. It is this type of information that is often unavailable from other study populations, such as those derived from a hospital-based case series. Nonetheless, caution should be taken when comparing our results across studies, or to the general population. Finally, the Long Island Breast Cancer Study Project was conducted in multiple hospitals; the treatment information was not as completed and detailed as an single institutional study. So we have limited ability to further investigate the predictive effect of BRCA1 methylation status.
In summary, we examined BRCA1 promoter methylation status and explored its relationship with clinicopathological factors and breast cancer survival. Our study, which is based on data drawn from a large population-based sample, is the first to report on the prognostic value of BRCA1 promoter methylation status in breast cancer in an epidemiologic study. Our results indicate that BRCA1 promoter methylation could be an important prognostic factor of breast cancer.
References
Landis SH, Murray T, Bolden S et al. (1999) Cancer statistics, 1999. CA Cancer J Clin 49:8–31
Russo J, Yang X, Hu YF et al (1998) Biological and molecular basis of human breast cancer. Front Biosci 3:D944–D960
Baylin SB, Herman JG, Graff JR et al (1998) Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res 72:141–196. doi:10.1016/S0065-230X(08)60702-2
Ehrlich M (2002) DNA methylation in cancer: too much, but also too little. Oncogene 21:5400–5413. doi:10.1038/sj.onc.1205651
Herman JG (1999) Hypermethylation of tumor suppressor genes in cancer. Semin Cancer Biol 9:359–367. doi:10.1006/scbi.1999.0138
Widschwendter M, Jones PA (2002) DNA methylation and breast carcinogenesis. Oncogene 21:5462–5482. doi:10.1038/sj.onc.1205606
Ralhan R, Kaur J, Kreienberg R et al (2007) Links between DNA double strand break repair and breast cancer: accumulating evidence from both familial and nonfamilial cases. Cancer Lett 248:1–17. doi:10.1016/j.canlet.2006.06.004
Miki Y, Swensen J, Shattuck-Eidens D et al (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266:66–71. doi:10.1126/science.7545954
Hopper JL (2001) Genetic epidemiology of female breast cancer. Semin Cancer Biol 11:367–374. doi:10.1006/scbi.2001.0392
Yang X, Yan L, Davidson NE (2001) DNA methylation in breast cancer. Endocr Relat Cancer 8:115–127. doi:10.1677/erc.0.0080115
Birgisdottir V, Stefansson OA, Bodvarsdottir SK et al (2006) Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res 8:R38. doi:10.1186/bcr1522
Butcher DT, Rodenhiser DI (2007) Epigenetic inactivation of BRCA1 is associated with aberrant expression of CTCF and DNA methyltransferase (DNMT3B) in some sporadic breast tumours. Eur J Cancer 43:210–219. doi:10.1016/j.ejca.2006.09.002
Pathology of familial breast cancer: differences between breast cancers in carriers of BRCA1 or BRCA2 mutations and sporadic cases (1997) Breast Cancer Linkage Consortium. Lancet 349:1505–1510. doi:10.1016/S0140-6736(96)10109-4
Robson ME, Chappuis PO, Satagopan J et al (2004) A combined analysis of outcome following breast cancer: differences in survival based on BRCA1/BRCA2 mutation status and administration of adjuvant treatment. Breast Cancer Res 6:R8–R17. doi:10.1186/bcr658
Stoppa-Lyonnet D, Ansquer Y, Dreyfus H et al (2000) Familial invasive breast cancers: worse outcome related to BRCA1 mutations. J Clin Oncol 18:4053–4059
Chappuis PO, Kapusta L, Begin LR et al (2000) Germline BRCA1/2 mutations and p27(Kip1) protein levels independently predict outcome after breast cancer. J Clin Oncol 18:4045–4052
Goffin JR, Chappuis PO, Begin LR et al (2003) Impact of germline BRCA1 mutations and overexpression of p53 on prognosis and response to treatment following breast carcinoma: 10-year follow up data. Cancer 97:527–536. doi:10.1002/cncr.11080
Moller P, Evans DG, Reis MM et al (2007) Surveillance for familial breast cancer: Differences in outcome according to BRCA mutation status. Int J Cancer 121:1017–1020. doi:10.1002/ijc.22789
Dietary reference intakes for folate, thiamin, riboflavin, niacin, vitamin B12, panthothenic acid, biotin, and choline (1998) National Academies Press, Washington, DC
Christman JK, Sheikhnejad G, Dizik M et al (1993) Reversibility of changes in nucleic acid methylation and gene expression induced in rat liver by severe dietary methyl deficiency. Carcinogenesis 14:551–557. doi:10.1093/carcin/14.4.551
Fowler BM, Giuliano AR, Piyathilake C et al (1998) Hypomethylation in cervical tissue: is there a correlation with folate status? Cancer Epidemiol Biomarkers Prev 7:901–906
Rampersaud GC, Kauwell GP, Hutson AD et al (2000) Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am J Clin Nutr 72:998–1003
Jacob RA, Gretz DM, Taylor PC et al (1998) Moderate folate depletion increases plasma homocysteine and decreases lymphocyte DNA methylation in postmenopausal women. J Nutr 128:1204–1212
Paz MF, Avila S, Fraga MF et al (2002) Germ-line variants in methyl-group metabolism genes and susceptibility to DNA methylation in normal tissues and human primary tumors. Cancer Res 62:4519–4524
Stern LL, Mason JB, Selhub J et al (2000) Genomic DNA hypomethylation, a characteristic of most cancers, is present in peripheral leukocytes of individuals who are homozygous for the C677T polymorphism in the methylenetetrahydrofolate reductase gene. Cancer Epidemiol Biomarkers Prev 9:849–853
Friso S, Choi SW, Girelli D et al (2002) A common mutation in the 5, 10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc Natl Acad Sci USA 99:5606–5611. doi:10.1073/pnas.062066299
Chen J, Gammon MD, Chan W et al (2005) One-carbon metabolism, MTHFR polymorphisms, and risk of breast cancer. Cancer Res 65:1606–1614. doi:10.1158/0008-5472.CAN-04-2630
Xu X, Gammon MD, Zhang H et al (2007) Polymorphisms of one-carbon metabolizing genes and risk of breast cancer in a population-based study. Carcinogenesis 28:1504–1509
Xu X, Gammon MD, Zeisel SH et al (2008) Choline metabolism and risk of breast cancer in a population-based study. FASEB J [Epub ahead of print]
Gammon MD, Neugut AI, Santella RM et al (2002) The Long Island breast cancer study project: description of a multi-institutional collaboration to identify environmental risk factors for breast cancer. Breast Cancer Res Treat 74:235–254. doi:10.1023/A:1016387020854
Cleveland RJ, Eng SM, Abrahamson PE et al (2007) Weight gain prior to diagnosis and survival from breast cancer. Cancer Epidemiol Biomarkers Prev 16:1803–1811. doi:10.1158/1055-9965.EPI-06-0889
Gammon MD, Santella RM, Neugut AI et al (2002) Environmental toxins and breast cancer on Long Island. I. Polycyclic aromatic hydrocarbon DNA adducts. Cancer Epidemiol Biomarkers Prev 11:677–685
Gaudet MM, Britton JA, Kabat GC et al (2004) Fruits, vegetables, and micronutrients in relation to breast cancer modified by menopause and hormone receptor status. Cancer Epidemiol Biomarkers Prev 13:1485–1494
McShane LM, Altman DG, Sauerbrei W et al (2006) REporting recommendations for tumor MARKer prognostic studies (REMARK). Breast Cancer Res Treat 100:229–235. doi:10.1007/s10549-006-9242-8
Zeisel SH, Mar MH, Howe JC et al (2003) Concentrations of choline-containing compounds and betaine in common foods. J Nutr 133:1302–1307
Hosmer DW (1999) Applied survival analysis : regression modeling of time to event data. Wiley, New York
Rosen EM, Fan S, Pestell RG et al (2003) BRCA1 gene in breast cancer. J Cell Physiol 196:19–41. doi:10.1002/jcp. 10257
Herman JG, Graff JR, Myohanen S et al (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 93:9821–9826. doi:10.1073/pnas.93.18.9821
Hilakivi-Clarke L (2000) Estrogens, BRCA1, and breast cancer. Cancer Res 60:4993–5001
Fan S, Wang J, Yuan R et al (1999) BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science 284:1354–1356. doi:10.1126/science.284.5418.1354
Fan S, Ma YX, Wang C et al (2001) Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene 20:77–87. doi:10.1038/sj.onc.1204073
Liebens FP, Carly B, Pastijn A et al (2007) Management of BRCA1/2 associated breast cancer: a systematic qualitative review of the state of knowledge in 2006. Eur J Cancer 43:238–257. doi:10.1016/j.ejca.2006.07.019
Yoshikawa K, Honda K, Inamoto T et al (1999) Reduction of BRCA1 protein expression in Japanese sporadic breast carcinomas and its frequent loss in BRCA1-associated cases. Clin Cancer Res 5:1249–1261
Perez-Valles A, Martorell-Cebollada M, Nogueira-Vazquez E et al (2001) The usefulness of antibodies to the BRCA1 protein in detecting the mutated BRCA1 gene. An immunohistochemical study. J Clin Pathol 54:476–480. doi:10.1136/jcp. 54.6.476
Al-Mulla F, Abdulrahman M, Varadharaj G et al (2005) BRCA1 gene expression in breast cancer: a correlative study between real-time RT-PCR and immunohistochemistry. J Histochem Cytochem 53:621–629. doi:10.1369/jhc.4A6544.2005
Wilson CA, Ramos L, Villasenor MR et al (1999) Localization of human BRCA1 and its loss in high-grade, non-inherited breast carcinomas. Nat Genet 21:236–240. doi:10.1038/6029
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by grants from the National Institutes of Health (CA109753 to JC; DK55865 to SZ) and in part by grants from Department of Defense (BC031746), National Cancer Institute and the National Institutes of Environmental Health and Sciences (UO1CA/ES66572, UO1CA66572, P30CA013696, P30ES09089 and P30ES10126); and by the University of North Carolina Clinical Nutrition Research Unit (DK56350) and Center for Environmental Health and Susceptibility (ES10126). Xu, X. is a recipient of the Predoctoral Traineeship Award (W81XWH-06-1-0298) of Department of Defense Breast Cancer Research Program.
Rights and permissions
About this article
Cite this article
Xu, X., Gammon, M.D., Zhang, Y. et al. BRCA1 promoter methylation is associated with increased mortality among women with breast cancer. Breast Cancer Res Treat 115, 397–404 (2009). https://doi.org/10.1007/s10549-008-0075-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-008-0075-5