
Abstract
Newborn screening for Fabry disease in Taiwan Chinese has revealed a high incidence of the late-onset GLA mutation IVS4 + 919G→A (∼1 in 1,500–1,600 males). We studied 94 adults with this mutation [22 men, 72 women; mean age: men 57.8 ± 6.0 (range 42–68), women 39.1 ± 14.1 years (range 19–82)]. Plasma α-galactosidase A activity assay was 10.4 ± 11.2% of normal in the men and 48.6 ± 19.5% of normal in the women. Echocardiography in 90 of the adults revealed left ventricular hypertrophy (LVH) in 19 (21%), including 14 of 21 men (67%) and 5 of 69 women (7%). Microalbuminuria, based on the urine albumin-to-creatinine ratio measured on at least two occasions, was present in 17 of 86 subjects (20%) (men: 5/20, 25%; women 12/66, 18%). At least one ocular manifestation consistent with Fabry disease was present in 41 of 52 subjects (79%) who underwent ophthalmologic examination, including 8 (15%) with conjunctival vessel tortuosity, 15 (29%) with cornea verticillata, 10 (19%) with Fabry cataract, and 34 (65%) with retinal vessel tortuosity. Among subjects over 40 years of age, men were more likely than women to have LVH [14/21 (67%) vs 5/25 (20%), p < 0.001]. Cardiovascular, renal and ocular abnormalities are highly prevalent in adult Taiwan Chinese subjects with the Fabry mutation IVS4 + 919G→A. Our findings contribute to the limited understanding of the course of this late-onset disease variant and underscore the need for close follow up in such patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fabry disease (MIM 301500) is an X-linked lysosomal storage disorder characterized by deficient α-galactosidase A (α-Gal A) activity, leading to progressive accumulation of globotriaosylceramide (GL-3) in the vascular endothelium of the skin, kidney, heart, and brain. It is a complex, multisystemic disorder characterized clinically by angiokeratomas, acroparesthesias, hypohydrosis, corneal opacities, gastrointestinal disturbances, progressive renal impairment, cardiomyopathy, and early stroke (Desnick et al. 2001). The estimated incidence of classic Fabry disease is 1 in 40,000–60,000 males in the general population (Desnick et al. 2001; Meikle et al. 1999). There is increasing recognition that manifestations in heterozygous females can range from no symptoms at all to abnormalities as severe as those in affected males (Deegan et al. 2006; Wang et al. 2007; Wilcox et al. 2008).
During the past decade, there have been reports of late-onset phenotypes of Fabry disease primarily involving the heart (Monserrat et al. 2007; Nakao et al. 1995; Sachdev et al. 2002), kidneys (Kotanko et al. 2004; Nakao et al. 2003; Tanaka et al. 2005) or cerebrovascular system (Rolfs et al. 2005). Patients with the cardiac variant lack the classic symptoms of Fabry disease and present with left ventricular hypertrophy (LVH), arrhythmias, or hypertrophic cardiomyopathy in the fifth to eighth decades of life. It has been suggested that 1–4% of men with LVH or hypertrophic cardiomyopathy have undiagnosed Fabry disease (Monserrat et al. 2007; Nakao et al. 1995; Sachdev et al. 2002). Patients with the renal variant develop proteinuria and may progress to end-stage renal disease, typically after 50 years of age. Screening by plasma α-Gal A activity has shown that 0.25–1% of men undergoing hemodialysis were identified with previously undiagnosed Fabry disease (Kotanko et al. 2004; Nakao et al. 2003; Tanaka et al. 2005). Ocular findings may suggest the diagnosis in some individuals, where progressive deposition of GL-3 in ocular structures may result in abnormalities at the level of the conjunctival vessels, cornea, lens, or retinal vessels (Nguyen et al. 2005; Orssaud et al. 2003; Sher et al. 1979; Sodi et al. 2007).
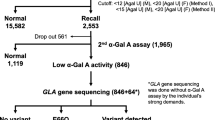
In Japan, an IVS4 + 919G→A splicing mutation has been reported in patients with the late-onset cardiac phenotype (Ishii et al. 2002). Lin et al. (2009) screened ∼57,000 newborn boys and found various Fabry mutations in ∼1 in 1,400, 83% of whom had the cardiac variant mutation IVS4 + 919G→A, for an incidence of ∼1 in 1,600. Hwu et al. (2009) screened ∼90,000 baby boys and found Fabry mutations in ∼1 in 1,250, 86% with IVS4 + 919G→A, an incidence of 1 in 1,500. This suggests a substantial incidence of the mutation in the Taiwanese population as a whole, but little is known about its phenotype in adult men and women. We previously looked for LVH in 20 maternal grandparents of babies carrying this mutation (Lin et al. 2009). The aim of the present study was to perform enzyme assays and assess clinical manifestations in a larger group of Taiwanese adults who carry the IVS4 + 919G→A mutation in order to help to delineate the natural history of late-onset Fabry disease associated with this genotype.
Materials and methods
Study design and subjects
Pedigree analysis was offered to the parents of the newborns with the IVS4 + 919G→A mutation who had been identified via the Fabry disease newborn screening program in Taiwan (Lin et al. 2009). Once families had consented to participate, parents, grandparents and other blood relatives underwent mutation analysis. Ninety-four adult subjects who were found to have the IVS4 + 919G→A mutation were enrolled in the current study. Informed written consent was obtained from each of the subjects. The study was approved by the ethics committee of Taipei Veterans General Hospital, Taipei, Taiwan.
Molecular, biochemical and clinical assessments
The α-Gal A exons and adjacent intronic and promoter regions were sequenced using standard techniques as described elsewhere (Shabbeer et al. 2005; Shabbeer et al. 2006). Plasma α-Gal A enzyme activity was measured according to the method described by Desnick et al. (1973) and expressed as numerical value and as the percentage of the mean in normals (i.e. 12.4 nmol/h/mL plasma; Desnick et al. 1973; Sheu et al. 1994).
A total of 94 subjects carried the mutation and were asked to undergo screening for cardiac, renal, and ocular abnormalities. However, it was up to the subjects to follow through with all instructions. All had at least one of the examinations.
Spot urine samples for measurement of albumin and creatinine levels were collected on at least two occasions to rule out confounding factors unrelated to Fabry disease. The ratio of concentrations of urinary albumin and creatinine expressed as mg/mmol was used to estimate the total daily albumin excretion. Microalbuminuria was defined as urinary albumin-to-creatinine ratio ≥2.0 mg/mmol for men and ≥2.8 mg/mmol for women on at least two occasions, based on the National Kidney Foundation's Kidney Disease Outcome Quality Initiative working group definition (National Kidney Foundation 2002).
Cardiac evaluation included electrocardiography and standard echocardiography. LVH was defined as left ventricular mass >259 g in men and >166 g in women (Levy et al. 1987).
Ophthalmological evaluation included slit lamp examination of the bulbar conjunctiva, cornea, lens and fundus.
Statistical analysis
The Statistical Package for Social Sciences (SPSS®) version 11.5 (SPSS, Chicago, IL) was used to analyze the plasma α-Gal A activity, as well as clinical manifestations and gender differences. Relationships between clinical findings and age, as well as clinical findings and plasma α-Gal A activity, were tested using Pearson correlation, and significance was tested using Fisher r–z transformations. Differences were considered to be statistically significant if the p value was less than 0.05.
Results
Among the 94 adult subjects with the IVS4 + 919G→A mutation, there were 22 men and 72 women. Their mean age (±SD, range) was 57.8 (6.0, 42-68) and 39.1 (14.1, 19-82) years, respectively (Fig. 1). Half the subjects were women under 40 years of age, and 22 men (23% of all subjects) and 25 women (27%) were older than 40.

Age distribution of adult subjects with late-onset Fabry mutation IVS4 + 919G→A (n = 94; 72 women, 22 men)
The plasma α-Gal A activity was analyzed in all subjects; the mean (±SD, % of normal) values were 1.29 (1.39, 10.4 ± 11.2) nmol/h/mL plasma for men and 6.03 (2.42, 48.6 ± 19.5) nmol/h/mL plasma for women (Fig. 2). There were no correlations (p > 0.05) between clinical findings and plasma α-Gal A activity in both men and women.

Residual α-galactosidase activity in women (n = 72) and men (n = 22) with late-onset Fabry mutation IVS4 + 919G→A. Activity is expressed as percentage of the normal mean (12.4 ± 2.25 nmol/h/mL plasma)
Echocardiographic examinations (n = 90) revealed LVH in 67% of 21 men assessed and in 7% of 69 women (Table 1). Men over 40 years were more likely to have LVH than women over 40 (67 and 20%, respectively; p < 0.001). LVH was more likely to occur in women over 40 than in women under 40 years of age (p < 0.005) and developed progressively (r = 0.588, p < 0.01).Microalbuminuria or higher levels of proteinuria were found in 17 subjects (20% of 86), including 5 men (25%) and 12 women (18%; Table 1). There was no significant difference between its presence in men (25%) and women (30%) older than 40 (p = 0.700), and between women older and younger than 40 (p = 0.06). However, (micro)albuminuria or proteinuria developed progressively in women (r = 0.281, p < 0.05).
At least one Fabry-related ocular manifestation was present in 41 subjects (79% of subjects examined); in 43% of men and 84% of women. Ocular abnormalities included retinal vessel tortuosity (29%, 71%, respectively), cornea verticillata (14%, 31%), Fabry cataract (0%, 22%) and conjunctival vessel tortuosity (14%, 16%) (Table 1, Fig. 3). There were no statistically significant differences between genders or age categories. The examinations were mostly performed by PKL (79%), whereas 21% of the subjects were examined by other ophthalmologists at the ophthalmology outpatient clinic.

Prevalence of individual abnormalities in men (n = 22) and women (n = 72) with late-onset Fabry mutation IVS4 + 919G→A
Discussion
The natural history of early-onset “classic” Fabry disease is well documented in the literature (Desnick et al. 2001; Zarate and Hopkin 2008). Cases of late-onset forms of Fabry disease have been reported but, to date, there are no reports of clinical findings in sizeable cohorts of patients. This is the first report describing the clinical features in both male and female patients carrying the IVS4 + 919G→A mutation in the GLA gene. We confirm a high prevalence of cardiovascular, renal, and ocular manifestations associated with Fabry disease in these Taiwan Chinese adult subjects, including women. Similar to the findings of Ishii et al. (2002) of ∼10% residual α-Gal A activity in lymphocytes from patients hemizygous for the IVS4 + 919G→A mutation, the mean enzyme activity in the 22 men in our study was 10.4% of normal. Our study is the first to report α-Gal A activities in heterozygous women with the IVS4 + 919G/A mutation, i.e. 48.6% of the normal mean (n = 72), and 14 women (19.4%) had normal α-Gal A enzyme activity (normal range: 7.9–16.9 nmol/h/mL plasma; Desnick et al. 1973; Sheu et al. 1994).
Among subjects over 40 years of age, 67% (14/21) of men and 20% (5/25) of women had LVH. The high incidence in men is consistent both with the study from Japan and our earlier investigation (Ishii et al. 2002; Lin et al. 2009). However, the present study also revealed a substantial number of women over 40 with the mutation who also had LVH. Our study was not designed to rule out other causes of LVH, so we cannot conclusively state that these individuals in fact had late-onset Fabry disease as a cause of their cardiac abnormality. However, we have previously found some cases of hypertrophic cardiomyopathy in which the patients carried this mutation (Lin et al. 2009). In a population with a relatively high incidence of this genotype, perhaps gene testing should be considered in patients who appear to have idiopathic hypertrophic cardiomyopathy.
Microalbuminuria was also relatively common in our subjects who were tested, one quarter of the men and nearly a third of the women over 40 years of age. As with LVH, it is possible that they may have had latent renal disease from other causes. But our study suggests that late-onset Fabry disease belongs in the differential diagnosis for these individuals.
Ocular findings are also among the early hallmarks of Fabry disease (Nguyen et al. 2005; Orssaud et al. 2003; Sher et al. 1979; Sodi et al. 2007) and readily detectable by slit lamp examination. Progressive deposition of GL-3 in ocular structures often leads to cornea verticillata (vortex keratopathy), changes of conjunctival and retinal vessels (dilatation, tortuosity, aneurysms) or lenticular changes (a “spoke-like” pattern at the level of the posterior capsule, usually referred to as “Fabry cataract”). Most women (84%) and less than half (43%) of men had at least one of these ocular manifestations. The prevalences of Fabry cataract (22%) and retinal vessel tortuosity (71%) among females were surprisingly high as compared to previous reports (Table 2). It remains to be elucidated if there is an ethnic predilection for these types of ocular abnormalities in the Taiwan Chinese Fabry patient population, or specifically in subjects with the IVS4 + 919G→A mutation. The prevalence of cornea verticillata was considerably lower (14% of men, 31% of women) as compared with the overall gender-specific prevalences of ∼75% reported by other groups (Table 2). This may support the belief that the IVS4 + 919G→A mutation is a rather mild pathogenic mutation.
Our findings demonstrate that the phenotype associated with this mutation cannot be accurately predicted from the genotype alone. As expected, men who were hemizygous for IVS4 + 919G→A had lower enzyme activity than did women who were heterozygous. Further investigations are needed to identify genes or other factors that modify the clinical expression of late-onset Fabry disease related to this mutation.
Enzyme replacement therapy has been used to treat Fabry disease and experience reveals that early medical intervention provides a better clinical outcome (Banikazemi et al. 2007; Weidemann et al. 2009; Wraith et al. 2008). This leaves little doubt that early detection and timely therapy are important for patients with classic Fabry disease. These paradigms are also applicable for patients with the late-onset mutation IVS4 + 919G→A as cardiovascular, renal and ocular manifestations are highly prevalent among these individuals. Our data contribute to the understanding of the clinical course of this late-onset variant and provide a rational for modification of current follow up and therapeutic intervention strategies.
Limitations
Fabry disease is an X-linked lysosomal storage disorder, but we had relatively few men in our sample. As subjects were selected based on the family pedigree of neonates found to have the IVS4 + 919G→A mutation, most were young mothers or maternal grandparents. Half were women younger than 40. It would be good to study larger groups of men, especially those younger than 40. Another limitation is the lack of complete data for all of our subjects, as they were not uniformly able to follow through on all the examinations.
Conclusion
This study documents a high prevalence of cardiovascular, renal, and ocular manifestations in Taiwan-Chinese adult subjects with Fabry mutation IVS4 + 919G→A. This hotspot mutation has been described as a “cardiac variant” mutation, but our data demonstrate that the clinical manifestations are not confined only to the heart. Although more detailed data on the longitudinal progression of the disease will be required, our findings will be helpful in determining the necessity and timing of therapeutic intervention with enzyme replacement therapy.
References
Banikazemi M, Bultas J, Waldek S et al (2007) Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med 146:77–86
Deegan P, Bähner AF, Barba-Romero MA, Hughes D, Kampmann C, Beck M (2006) Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet 43:347–352
Desnick RJ, Allen KY, Desnick SJ, Raman MK, Bernlohr RW, Krivit W (1973) Fabry's disease: enzymatic diagnosis of hemizygotes and heterozygotes. Alpha-galactosidase activities in plasma, serum, urine, and leukocytes. J Lab Clin Med 81:157–171
Desnick RJ, Ioannou YA, Eng CM (2001) Alpha-Galactosidase a deficiency: fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds); Childs B, Kinzler KW, Vogelstein B (assoc eds). The metabolic and molecular bases of inherited disease, 8th edn. McGraw-Hill, New York, pp 3733–3774
Hwu WL, Chien YH, Lee NC et al (2009) Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936 + 919G>A (IVS4 + 919G>A). Hum Mutat 30:1397–1405
Ishii S, Nakao S, Minamikawa-Tachino R, Desnick RJ, Fan JQ (2002) Alternative splicing in the alpha-galactosidase A gene: increased exon inclusion results in the Fabry cardiac phenotype. Am J Hum Genet 70:994–1002
Kotanko P, Kramar R, Devrnja D et al (2004) Results of a nationwide screening for Anderson-Fabry disease among dialysis patients. J Am Soc Nephrol 15:1323–1329
Levy D, Savage DD, Garrison RJ, Anderson KM, Kannel WB, Castelli WP (1987) Echocardiographic criteria for left ventricular hypertrophy: the Framingham Heart Study. Am J Cardiol 59:956–960
Lin HY, Chong KW, Hsu JH et al (2009) High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ Cardiovasc Genet 2:450–456
Meikle PJ, Hopwood JJ, Clague AE, Carey WF (1999) Prevalence of lysosomal storage disorders. JAMA 281:249–254
Monserrat L, Gimeno-Blanes JR, Marín F et al (2007) Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 50:2399–2403
Nakao S, Takenaka T, Maeda M et al (1995) An atypical variant of Fabry's disease in men with left ventricular hypertrophy. N Engl J Med 333:288–293
Nakao S, Kodama C, Takenaka T et al (2003) Fabry disease: detection of undiagnosed hemodialysis patients and identification of a "renal variant" phenotype. Kidney Int 64:801–807
National Kidney Foundation (2002) K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 39:S1–S266
Nguyen TT, Gin T, Nicholls K, Low M, Galanos J, Crawford A (2005) Ophthalmological manifestations of Fabry disease: a survey of patients at the Royal Melbourne Fabry Disease Treatment Centre. Clin Experiment Ophthalmol 33:164–168
Orssaud C, Dufier J, Germain D (2003) Ocular manifestations in Fabry disease: a survey of 32 hemizygous male patients. Ophthalmic Genet 24:129–139
Rolfs A, Böttcher T, Zschiesche M et al (2005) Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet 366:1794–1796
Sachdev B, Takenaka T, Teraguchi H et al (2002) Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 105:1407–1411
Shabbeer J, Robinson M, Desnick RJ (2005) Detection of alpha-galactosidase a mutations causing Fabry disease by denaturing high performance liquid chromatography. Hum Mutat 25:299–305
Shabbeer J, Yasuda M, Benson SD, Desnick RJ (2006) Fabry disease: identification of 50 novel alpha-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations. Hum Genomics 2:297–309
Sher NA, Letson RD, Desnick RJ (1979) The ocular manifestations in Fabry's disease. Arch Ophthalmol 97:671–676
Sheu SS, Chan LP, Liao SC et al (1994) Fabry's disease: clinical, pathologic and biochemical manifestations in two Chinese males. Zhonghua Yi Xue Za Zhi 54:368–372
Sodi A, Ioannidis AS, Mehta A, Davey C, Beck M, Pitz S (2007) Ocular manifestations of Fabry's disease: data from the Fabry Outcome Survey. Br J Ophthalmol 91:210–214
Tanaka M, Ohashi T, Kobayashi M et al (2005) Identification of Fabry's disease by the screening of alpha-galactosidase a activity in male and female hemodialysis patients. Clin Nephrol 64:281–287
Wang RY, Lelis A, Mirocha J, Wilcox WR (2007) Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med 9:34–45
Weidemann F, Niemann M, Breunig F et al (2009) Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation 119:524–529
Wilcox WR, Oliveira JP, Hopkin RJ et al (2008) Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab 93:112–128
Wraith JE, Tylki-Szymanska A, Guffon N et al (2008) Safety and efficacy of enzyme replacement therapy with agalsidase beta: an international, open-label study in pediatric patients with Fabry disease. J Pediatr 152:563–570
Zarate YA, Hopkin RJ (2008) Fabry's disease. Lancet 372:1427–1435
Acknowledgements
We would like to express our sincere thanks to Dr. Hans Ebels for his critical review and valuable comments and Ms. Tsai-Feng Ho for her professional assistance in biostatistics. This work was supported by Taipei Veterans General Hospital (V99C1-147).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by: Olaf Bodamer
Competing interest: None declared.
Rights and permissions
About this article
Cite this article
Lin, HY., Huang, CH., Yu, HC. et al. Enzyme assay and clinical assessment in subjects with a Chinese hotspot late-onset Fabry mutation (IVS4 + 919G→A). J Inherit Metab Dis 33, 619–624 (2010). https://doi.org/10.1007/s10545-010-9166-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-010-9166-7