
Abstract
The ubiquitin–proteasome system (UPS) is indispensable to the protein quality control in eukaryotic cells. Due to the remarkable clinical success of using proteasome inhibitors for clinical treatment of multiple myeloma, it is anticipated that targeting the UPS upstream of the proteasome step be an effective strategy for cancer therapy. Deubiquitinases (DUB) are proteases that remove ubiquitin from target proteins and therefore regulate multiple cellular processes including some signaling pathways altered in cancer cells. Thus, targeting DUB is a promising strategy for cancer drug discovery. Previously, we have reported that metal complexes, such as copper and gold complexes, can disrupt the UPS via suppressing the activity of 19S proteasome-associated DUBs and/or of the 20S proteasomes, thereby inducing cancer cell death. In this study, we found that cadmium pyrithione (CdPT) treatment led to remarkable accumulation of ubiquitinated proteins in cultured cancer cells and primary leukemia cells. CdPT potently inhibited the activity of proteasomal DUBs (USP14 and UCHL5), but slightly inhibited 20S proteasome activity. The anti-cancer activity of CdPT was associated with triggering apoptosis via caspase activation. Moreover, treatment with CdPT inhibited proteasome function and repressed tumor growth in animal xenograft models. Our results show that cadmium-containing complex CdPT may function as a novel proteasomal DUB inhibitor and suggest appealing prospects for cancer treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
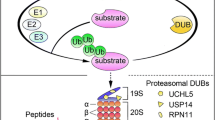
Protein homeostasis or proteostasis is pivotal to cellular processes that are essential to the fate and growth of cancer cells. The ubiquitin–proteasome system (UPS) is a highly regulated pathway responsible for the degradation of more than 80% of cellular proteins, including the regulators and participants of cell cycle progression, DNA damage responses and repair, and cell death (Adams 2003; Ciechanover 1994). In contrast to normal cells, cancer cells usually possess higher proteasome activities and are more sensitive to proteasome inhibition. Therefore, the UPS has attracted increasingly great attention from the search for new targets for cancer therapy (Adams 2004; Kazi et al. 2014; Wu et al. 2010). A protein to be degraded by the UPS is first covalently conjugated to a chain of ubiquitin proteins. The poly-ubiquitin chain then marks the substrate protein for recognition and degradation by the 26S proteasome, in which the 19S regulatory particle plays an indispensable role by binding to the ubiquitinated protein, removing and recycling the ubiquitin, and unfolding the substrate protein to channel it into the 20S proteasome where the peptide chain cleavage takes place. The 20S proteasome harbors three major protease activities: chymotrypsin-like (CT-L) activity, trypsin-like (T-L) activity, and caspase-like (C-L) activity, that cleaves following hydrophobic, basic, and acidic residues, respectively (Ciechanover 1994; Ho et al. 2007). The significance of disrupting UPS-mediated proteolysis is corroborated by the successful treatment of some of the hematologic malignancies with proteasome inhibitors (e.g., Bortezomib, Carfilzomib) in the clinic. However, development of resistance to the current proteasome inhibitors and the poor efficacy of the proteasome inhibitors in treating solid tumors warrant the search for new strategies to disrupt protein homeostasis (Anderson et al. 2015; Dou and Li 1999).
Deubiquitinases (DUB) have fundamental roles in the UPS through their ability to specifically deconjugate ubiquitin from targeted proteins. The human genome encodes for approximately 100 DUBs, of which only three reside in or directly interact with the 19S regulatory particle of the 26S proteasome: POH1, USP14 and UCHL5 (D’Arcy et al. 2011). It is believed that POH1 cuts at the base of a ubiquitin chain and promotes translocation of the substrate into the 20S proteasome for degradation. In contrast, USP14 and UCHL5 are cysteine proteases that trim ubiquitin chains from the distal end, thereby shortening the chains rather than cleaving them en bloc (Koulich et al. 2008; Verma et al. 2002). Therefore, as important as ubiquitin ligases or the proteasome proper, the activity of DUBs also controls the rate of degradation, subcellular distribution and activation of proteins that regulate the life and death of cells. Thus, DUBs, the proteasome-associated DUBs in particular, are rapidly emerging as potential therapeutic targets for cancer therapy (D’Arcy et al. 2011; D’Arcy and Linder 2012; Fraile et al. 2012; Wei et al. 2015).
Presently, there is a large interest in the research of metal complexes as proteasome inhibitors (Liu et al. 2015; Verani 2012). Several copper chelating agents, such as disulfiram, pyrrolidine dithiocarbamate, clioquinol, and pyrithione (PT), can bind copper and the resultant new complexes had proven to effectively inhibit the 20S proteasome and induce cell death in cancer cells (Chen et al. 2006; Daniel et al. 2005; Liu et al. 2014b). Some of these metal compounds showed much less inhibitory effects against purified 20S proteasomes than against cellular 26S proteasomes. It has been proposed that inhibition of DUBs in the 19S proteasome is possibly responsible for the anti-tumor effect of these metal complexes (Cvek et al. 2008; Liu et al. 2014b; Skrott and Cvek 2012). We have recently found that gold (I) compound Auranofin induces cancer cell death via inhibition of 19S proteasome-associated DUBs (USP14 and UCHL5) but not the 20S proteasome (Liu et al. 2014a). In the present study, we have discovered that cadmium-containing compound CdPT targets proteasomal DUBs, and inhibits tumor growth in vitro and in vivo.
Materials and methods
Materials
The reagents used in this study and their sources were: K2CdCl4 (Cd2+), pyrithione (PT), CDDP, NEM and rhodamine-123 (Sigma-Aldrich, St. Louis, MO, USA). CdPT was synthesized in our lab; Velcade (BD Biosciences, San Jose, CA, USA); b-AP15, purified 20S and 26S proteasomes, Suc-LeuLeu-Val-Tyr-aminomethylcoumarin (Suc-LLVY-AMC), Z-Leu-Leu-Glu-AMC (Z-LLE-AMC), Boc-Leu-Arg-Arg-AMC (Boc-LRR-AMC), Ubiquitin-AMC (U550), K48-linked tetra-ubiquitin, HA-Ubiquitin-Vinyl Sulfone (HA-Ub-VS); and Enhanced chemiluminescence (ECL) reagents (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Antibodies were obtained from the following sources: antiubiquitin (P4D1), anti-GFP (B-2) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-PARP, anti-caspase 3 (8G10), anti-K48-linkage specific polyubiquitin (D9D5), anti-p21 Waf1/Cip1 (DCS60) (Cell Signaling Technology, Beverly, MA, USA); and anti-GAPDH, anti-HA, anti-cleaved caspase 3 (Bioworld Technology, Inc., St Louis Park, MN, USA).
Cell culture
A549 and cisplatin-resistant A549/DDP cell lines were provided by Dr. Z. He (Cancer Hospital and Cancer Research Institute of Guangzhou Medical University). K562 (chronic myelogenous leukemia cells), U266 (myeloma cells), HEK293 (human embryonic epithelial cells) cell lines were purchased from American Type Culture Collection (Manassas, VA, USA), and has been recently authenticated. A549, K562 and U266 cell lines were cultured in RPMI 1640 medium (Gibco-Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS). A549/DDP cells were routinely maintained in the same medium but in the presence of 1.5 μg ml−1 cisplatin, which was removed before experiments were started by a wash-out period of 2–3 days. HEK-293 cells stably harboring GFPu were created as we previously described (Huang et al. 2010), and cultured in DMEM medium with high glucose supplemented with 10% FBS.
Cell viability assay
The cell viability assay kit (CellTiter 96 Aqueous One Solution reagent) was purchased from Promega (Shanghai, China). Briefly, 10,000 cells per well were seeded in a 96-well plate and treated with CdPT or cisplatin. 3 h before the cell culture termination, MTS solution (20 μl) was added to each well of the 96-well plate. The absorbance density was recorded at wavelength 490 nm using a plate reader (Varioskan Flash 3001, Thermo, Waltham, MA, USA). IC50 values were calculated.
Cell death assay
Apoptosis was assessed by flow cytometry using Annexin V-FITC/PI apoptosis detection kit (Keygen Biotechnology, Nanjing, China). Cultured cells were harvested, washed three times with the binding buffer, and then incubated in the working solution (500 μl binding buffer with 5 μl Annexin V-FITC) for in the dark 15 min; Immediately before analysis by flow cytometry, 5 μl PI was added to each sample. In addition, Annexin V-FITC/PI staining was also performed as described but in situ. An inverted fluorescence microscope equipped with a digital camera (Axio Obsever Z1, Zeiss, Jena, Germany) was used to image the double stained cells.
Mitochondrial membrane potential
The integrity of mitochondrial membrane was assessed by flow cytometry using rhodamine-123 staining. Cells were treated with various concentrations of CdPT, and then stained with rhodamine-123 (1 μM) in phosphate buffer saline (PBS) for 1 h at 37 °C. Following the staining, unbounded dye was removed by washing the cells three times with PBS and harvested for flow cytometric analysis.
Western blot analysis
Whole-cell lysates were prepared using RIPA lysis buffer. Western blotting analysis were performed as previously described (Chen et al. 2014). In brief, equal protein amounts were separated on 12% SDS-PAGE and electrically transferred onto a PVDF membrane. The membranes were incubated with the primary antibodies at 4 °C overnight and then incubated with appropriate horseradish peroxidase-conjugated secondary antibodies for 1 h. After washing, the bound secondary antibodies on the PVDF membrane were detected using the ECL detection reagents (Santa Cruz Biotechnology) followed by exposure to X-ray films (Kodak, Rochester, NY, USA).
20S proteaome peptidase activity assay
This was performed as we previously reported (Zhao et al. 2016a). Proteasome activity was measured using fluorogenic substrates Suc-LLVYAMC, Z-LLE-AMC and Boc-LRR-AMC for chymotrypsinlike, caspase-like and trypsin-like activity, respectively. Purified human 20S proteasomes were treated with CdPT or Velcade for 30 min, and then incubated with the corresponding fluorogenic substrate for 1 h at 37 °C. Fluorescence intensity of each sample was detected by a microplate reader (Varioskan Flash 3001, Thermo) at 350 nm excitation and 438 nm emission.
DUB activity assay
This was performed as we previously reported (Zhao et al. 2016a). Briefly, we incubated A549 cell lysate (5 μg) or purified 26S proteasome (25 nM) with CdPT or NEM in DUB buffer (25 mM Tris–HCl (pH 7.4), 20 mM NaCl, 5 mM MgCl2 and 200 μM ATP) for 30 min, and then Ub-AMC substrate was added to the reaction mixture. AMC was released from substrate cleavage mediated by DUB. The fluorescence intensity was temporally recorded with a microplate reader (Varioskan Flash 3001, Thermo).
Ubiquitin chain disassembly
This was performed as we previously reported (Zhao et al. 2016a). 25 nM purified 26S proteasomes were treated with CdPT, b-AP15 or TPEN for 10 min and then incubated with purified K48-linked tetra-ubiquitin chains (500 ng) in DUB buffer (25 mM Tris–HCl (pH 7.4), 20 mM NaCl, 5 mM MgCl2 and 200 μM ATP) for 30 min at 37 °C. The mixture was then fractionated using SDS-PAGE, transferred to PVDF membrane and immunodetected using anti-ubiquitin antibodies to assess for the extent of chain disassembly.
DUB active-site-directed labeling assays
This was performed as we previously reported (Zhao et al. 2016a). 25 nM purified 26S proteasomes were treated with CdPT or b-AP15 for 10 min and then incubated with HA-UbVS (400 nM) in DUB buffer (25 mM Tris–HCl (pH 7.4), 20 mM NaCl, 5 mM MgCl2 and 200 μM ATP) for 1 h at 37 °C. Followed by boiling in the reducing loading buffer, the sample was separated by SDS-PAGE and transferred to PVDF membranes, DUBs labeled by HA-UbVS were immunodetected using anti-HA antibodies.
Computational modeling
To predict the binding of CdPT toward the DUBs (USP14 and UCHL5), molecular docking studies were performed with CDOCKER protocol of Discovery Studio 2.0 [Accelrys Software Inc. (2007)] as we reported previously (Liu et al. 2014b). The crystallographic structures of USP14 and UCHL5 were directly downloaded from the Protein Data Bank (PDB IDs: 2AYO and 3RIS). After removing irrelevant components, hydrogen atoms were added and their positions were minimized with a 0.01 kcal/mol/Å root mean square gradient by using the all-atom CHARMm forcefield and the Adopted Basis Newton–Raphson (NR) Algorithm. In addition, taking into account the possible hydrolysis of compound CdPT (L1) in certain physiological conditions, the hydrolysate (L2) was selected as the docking ligand. The geometry structure of compound L2 was optimized using the DFT calculations at the B3LYP/LANL2DZ level to obtain NPA charges by using the Gaussian 03 [Revision D.01, Gaussian, Inc., Wallingford, CT (2004)]. During the whole docking process, the two proteins were rigid, while the ligand L2 was flexible. The Input Site Spheres of 12 Å radius were centered on each active pocket of USP14 and UCHL5, with (x, y, z) = (38.12, 84.32, 6.61) and (− 9.40, 6.57, 61.54), respectively. The conformation corresponding to the lowest CDOCKER Interaction Energy was selected as the most probable binding conformation. All parameters used in calculation were default except for those explained.
Animals and xenograft tumor mouse model
All animal care and experimental procedures were approved by the Guangzhou Medical University Institutional Animal Care and Use Committee. The mice were obtained from Guangdong Laboratory Animal Monitoring Institute (SCXK2008-2002). Male nude Balb/c mice (5-week-old) were housed in an specific pathogen free (SPF) facility with a 12 h light–dark cycle, with ad libitum access to food and water. The mice were injected subcutaneously with approximately 1 × 107 of A549 cells. 72 h after inoculation, the mice were randomly assigned to vehicle (10% DMSO, 30% Cremophor EL and 60% normal saline) and CdPT treatment groups with 6 animals each. Animals of CdPT treatment group received 2.5 mg/kg/day injection ip every day for 15 days. Tumor volumes were recorded and calculated as we previously described (Shi et al. 2014).
Immunohistochemistry
Formalin-fixed and paraffin embedded sections (4 μm) from xenografts were immunostained using the MaxVision Kit (Maixin Biol, Fuzhou, China) according to the manufacturer’s instructions. After overnight incubation with primary antibodies as indicated, tissue sections were incubated with 50 μl MaxVision reagent. Color was developed with 0.05% diaminobenzidine and 0.03% H2O2 in 50 mM Tris–HCl (pH 7.6), and then counterstained with hematoxylin. Negative control was also included for each xenograft specimen by using pre-immune serum instead of the primary antibodies.
Isolation and culture of human peripheral blood monocytes
Peripheral blood samples of six healthy control individuals and bone marrow samples of six acute myeloid leukemia (AML) patients were obtained from Guangzhou Blood Center and Guangzhou First Municipal People’s Hospital, respectively. The study was approved by the Ethics Committee of Guangzhou Medical University with the prior permission of the volunteers. Mononuclear cells were isolated from the samples by density gradient centrifugation using Ficoll-Paque (Pharmacia, Uppsala, Sweden) density gradient and then cultured in RPMI 1640 medium supplemented with 15% FBS.
Data analysis
All the results were expressed as mean ± SD. where applicable. GraphPad Prism 5.0 software GraphPad Software, San Diego, CA, USA) was used for statistical analysis. Differences between two groups were evaluated for statistical significance using two-tailed Student’s t test. P value of < 0.05 was considered statistically significant.
Results
CdPT induces apoptosis in cultured cancer cells
We first tested the effects of CdPT on the viability of different human cancer cells. MTS assay was employed to assess cell viability after CdPT treatment for 24 h. As presented in Fig. 1, CdPT treatment led to decreases in the cell viability of a panel of cancer cells (A549, A549/DDP, K562 and U266) in a dose-dependent manner and displayed a greater efficacy than cisplatin (CDDP). A549 cells are sensitive, whereas A549/DDP cells are resistant to cisplatin. Notably, we discovered that CdPT was able to partially overcome the cisplatin resistance of A549/DDP cells, with the IC50 values of 2.15 and 3.57 μM in A549 and A549/DDP cells, respectively.

CdPT induced cytotoxicity in cancer cell lines. A549, A549/DDP, K562 and U266 cells were exposed to CdPT or CDDP in various concentrations for 24 h and then were subject to MTS assay. Data from three biological repeats are presented. Mean ± SD. (n = 3)
Next, we studied the ability of CdPT to induce cell death. We exposed the cancer cells to increasing concentrations of CdPT for 24 h and analyzed cell death using flow cytometry (A549 and U266 cells, Fig. 2a) or fluorescence microscopy (A549/DDP and K562 cells, Fig. 2b) following Annexin V-PI staining. We observed that CdPT-induced cell death was primarily in the form of apoptosis. Since mitochondria are known to be the center for the control of apoptosis (Marchetti et al. 1996), we examined the potential involvement of mitochondria in this setting. We found that mitochondrial membrane potential was decreased in A549 cells after CdPT treatment (Fig. 2c), indicating that CdPT treatment causes the disruption of the integrity of mitochondrial membranes. Consistent with activation of caspases by CdPT, the cleavage forms of PARP and caspase 3 in A549, A549/DDP and K562 cells were obviously increased after CdPT treatment (Fig. 2d). These findings indicate that CdPT induces caspase activation and apoptosis in the tested cancer cells.

Apoptosis induction by CdPT in cancer cell lines. A549 and U266 cells (a), or A549/DDP and K562 cells (b) were exposed to CdPT at the indicated dose for 24 h, then the cells undergoing apoptosis was investigated with flow cytometry (a) or imaged under a fluorescent microscope (b) after the cells were stained with Annexin V-FITC and PI (A/P). The extent of cell apoptosis was summarized in the associated bar graph (a). Mean ± SD. (n = 3). *P < 0.05 versus control. c CdPT induces downregulation of mitochondrial membrane potential. A549 cells were treated with 1.25, 2.5 and 5.0 μM CdPT for 12 h, and mitochondrial membrane potential were detected by rhodamine-123 (Rh123) staining followed by flow cytometry. The extent of mitochondrial membrane potential decrease was summarized in the bar graph below. Mean ± SD. (n = 3). *P < 0.05 versus control. d CdPT induces cleavage of PARP and caspases-3 activation in cancer cells. A549, A549/DDP and K562 cells were treated with the indicated concentrations of CdPT for 24 h; PARP, caspase 3 and cleaved caspase3 were analyzed with western blotting. GAPDH was used as a loading control. c-caspase-3: cleaved caspase3
CdPT triggers accumulation of ubiquitinated proteins
Several metal complexes have been shown to inhibit proteasome activities and demonstrate valuable effects as antineoplastic agents (Chen et al. 2014; Frezza et al. 2010; Verani 2012). Thus, we determined the impact of CdPT treatment on the level of endogenous ubiquitin-conjugated proteins which is a commonly used indicator of UPS functioning. We found striking increases of ubiquitin conjugates in CdPT-treated A549, A549/DDP and K562 cells; the increases of ubiquitinated proteins by CdPT are CdPT dose-dependent and the treatment time-dependent (Fig. 3a, b). Additionally, we also tested the effect of CdPT on the protein expression of a degron CL1-fused green fluorescence protein (GFPu), a well-established surrogate substrate of the UPS, stably expressed in clonal HEK-293 cells (Huang et al. 2010). Velcade (Vel, Bortezomib) is a bona fide inhibitor of 20S proteasome activities (Paramore and Frantz 2005). We found that both CdPT (2.5 and 5 μM) and Velcade (50 nM) could induce dramatic accumulations of GFPu proteins (Fig. 3c and d) and ubiquitinated proteins (Fig. 3d) in the GFPu-HEK-293 cells.

CdPT inhibits proteasome function in cultured cancer cells. a and b CdPT induces accumulation of ubiquitinated proteins. A549, A549/DDP and K562 cells were treated with CdPT at the indicated various concentrations for 24 h (a) or with the indicated concentrations for indicated times (12, 18, 24 h) (b), and then ubiquitinated proteins (Ub-prs.) were detected using western blot analyses. GAPDH was used as a loading control. c and d CdPT accumulates GFPu, a surrogate proteasome substrate created by carboxyl fusion of an enhanced green fluorescence protein with degron CL1. GFPu-HEK-293 cells, a clonal HEK-293 cell line stably transfected with GFPu, were treated with CdPT (2.5 μM) or Velcade (50 nM), the fluorescent GFPu images are shown in panel (c). GFPu-HEK293 cells were treated with the indicated concentrations of CdPT or Velcade (Vel, 50 nM) for 12 h, and then ubiquitinated proteins and GFP protein were detected with western blotting (d). GAPDH was used as a loading control. e Effect of CdPT on 20S proteasomes. Purified 20S proteasomes were treated with CdPT at the indicated doses in a Tris reaction system (pH 7.4). Velcade (Vel, 50 nM) was used as positive control. Thechymotrypsin-like (CT-like), caspase-like (T-like) and trypsin-like (T-like) activities of the 20S proteasomes were measured using specific fluorogenic substrates Suc-LLVYAMC, Z-LLE-AMC and Boc-LRR-AMC, respectively. Mean ± SD. (n = 3)
In general, ubiquitinated proteins are degraded by the 26S proteasome, which is composed of the 20S proteasome capped at one or both ends by 19S regulatory particles (Ciechanover 1994; Coux et al. 1996). To delineate the target of CdPT in the UPS, we measured the peptidase activities of purified 20S proteasomes after CdPT treatment. We found that CdPT only at high doses inhibits the 20S proteasome enzymatic activities, with the IC50 of 7.48 μM (CT-like activity), 8.92 μM (T-like activity) and 18.89 μM (C-like activity) (Fig. 3e). However, Velcade inhibited only the CT-like and C-like activities, but not the T-like activity, which is consistent with previous reports (Kisselev et al. 2006).
CdPT has a high affinity for USP14 and UCHL5
Given that for CdPT to directly inhibit 20S proteasome activities, a dose much higher than its effective doses to induce the accumulation of endogenous ubiquitinated proteins or cell death in the cultured cells is required, we hypothesized that CdPT may potentially act on other steps of the UPS in addition to direct inhibition of the 20S proteasome, for example, by targeting the DUB activity of the 19S proteasome. N-ethylmaleimide (NEM) is a broad-spectrum inhibitor of cysteine proteases (Mollah et al. 2007) and most DUBs (e.g., USP14 and UCHL5) are cysteine proteases; hence, NEM can inhibit these DUBs and were thus used as a positive control for DUB inhibition. We found that the DUB activities in cell lysates were moderately inhibited by CdPT (0.5, 1.0 and 2.0 μM) in a dose-dependent fashion (Fig. 4a) but CdPT at all the three tested doses (0.5, 1.0 and 2.0 μM) completely inhibited the DUB activities in the purified 26S proteasomes (Fig. 4b). Next, we tested the effect of CdPT on the proteasomal cleavage of tetraubiquitin chains (Ub4). b-AP15 has been reported as an inhibitor of USP14 and UCHL5, two DUBs that can be found in association with the 19S proteasome; POH1, the stoichiometric DUB subunit of the 19S proteasome, is sensitive to the zinc chelator TPEN (D’Arcy et al. 2011). We found that CdPT, b-AP15 or TPEN efficiently prevented the 26S proteasome from dissembling Ub4 in vitro (Fig. 4c). Using a competition binding assay between CdPT and HA-Ub-vinyl sulfone (HA-UbVS), we found that CdPT treatment (5, 50 and 100 μM) could reduce the binding of HA-UbVS to USP14 and UCHL5 (Fig. 4d). Taken together, these results consistently support that CdPT is an inhibitor of proteasomal DUBs (USP14 and UCHL5).

CdPT inhibits 26S proteasomal DUB activities. a Effects of CdPT on Ub-AMC cleavage by the DUBs in cell lysates. Cell lysate was treated with CdPT (0.5, 1.0, 2.0 μM) or NEM (2 mM) and then the DUB activity at different times was recorded by using the Ub-AMC substrate. The experiment was repeated three times and all yielded similar results. DUB activity was expressed as fold relative fluorescence units (RFU) difference over time. b Effects of CdPT on Ub-AMC cleavage by 26S proteasomes. Purified 26S proteasomes were treated as descibed in panel A, and then DUB activity was kinetically detected. c CdPT inhibits ubiquitin chain disassembly. K48-linked ubiquitin tetramers (Ub4) was incubated with 26S proteasomes in the absence or presence of CdPT (5, 50 and 100 μM) for 30 min, and then the product was subject to western blot analysis for ubiquitin protein species. b-AP15 (50 μM) and TPEN (250 μM) were used as positive controls. d Active-site-directed labeling of proteasomal DUBs. 26S proteasomes were treated with CdPT (5, 50 and 100 μM) and then labeled with HA-UbVS and resultant products were subject to western blot analysis for HA. b-AP15 (50 μM) was used as positive control
Both metal ion Cd2+ and ligand PT inhibit proteasomal DUB
Figure 5a depicts the interacting modes of compound L2 (the mediated product of CdPT) with the DUB active sites of USP14 and UCHL5. It is discernible that, in active sites, the Cd atom of compound L2 can form networks of two coordination bonds with the side chains of Asp451, while the S atom and O atom form two weak hydrogen bonds with His434 and Cys113 on USP14, respectively. Similarly to USP114, the catalytic core of UCHL5 can be blocked by CdPT as well. Three coordination bonds can be formed between the Cd atom of CdPT and the side chains of Gln82, His164 and Cys88 in UCHL5; meanwhile, the S atom and O atom of CdPT may respectively form two weak hydrogen bonds with Gln91 and Cys88 of UCHL5. Due to these strong interactions of compound L2 with the catalytic cores of these 19S proteasome-DUBs, it is predicted that the catalytic activity of these DUBs will be impaired in the presence of CdPT.

Effects of EDTA on CdPT-induced proteasome inhibition and apoptosis. a Computational molecular docking of CdPT with USP14 and UCHL5. The following data are shown: the structure of CdPT (L1); the structure of CdPT intermediate (L2); the binding modes of compound L2 at the active site of USP14 and UCHL5. b Effects of PT, CdPT or Cd2+ on Ub-AMC cleavage by 26S proteasomes. Purified 26S proteasomes were treated with the indicated doses (μM) of PT, CdPT or Cd2+, and then DUB activity was kinetically detected. Cd2+: K2CdCl4. c A549 cells were incubated with 5 μM CdPT in the absence or presence of 100 μM EDTA for 12 h, and then ubiquitinated proteins (Ub-prs.) and PARP proteins were detected using western blotting. GAPDH was used as a loading control. d K562 cells were treated as described in panel C, then the percentage of cell apoptosis was determined with Annexin V/PI staining followed by flow cytometry
To verify the contribution of Cd2+ or the chelator PT to the proteasome inhibition action of the CdPT complex, we tested the efficacy of Cd2+, PT, and CdPT in inhibiting the in vitro DUB activities of purified 26S proteasomes. We found that 0.63 μM PT or CdPT, rather than 0.63 μM Cd2+, obviously inhibited the DUB activities. Interestingly, 1.25 and 2.5 μM Cd2+, compared with CdPT or PT, had relatively weak inhibition on the DUB activities (Fig. 5b). Next, the importance role of Cd2+ was illustrated by the treatment of a powerful metal chelating agent EDTA. The addition of EDTA completely abrogated CdPT-induced ubiquitinated protein accumulation and PARP cleavage in A549 cells (Fig. 5c), or cell apoptosis in K562 cells (Fig. 5d). These results confirm that cadmium ion plays some roles in the inhibition of DUBs by CdPT but PT is the major determinant for the potency of CdPT.
The effect of CdPT on tumor growth in vivo
We next examined the in vivo anti-tumor effect of CdPT using A549 xenograft nude mouse. We observed that the dynamic tumor growth (Fig. 6a) and the tumor weights (Fig. 6b) were significantly restrained in the CdPT treatment group compared with the vehicle group. During the testing period, body weight gradually increased in the control group while kept relatively stable in CdPT-treated group (Fig. 6c). Our immunohistochemistry examination of the xenograft tissue samples revealed that compared with the vehicle treated groups, the CdPT-treated group showed markedly increases in the levels of total and K48-linked ubiquitin-conjugated proteins, tumor suppressor protein p21, both are proteasomal substrates, as well as the cleaved form of caspase 3 proteins (Fig. 6d). Together, these data demonstrate that CdPT is capable of effectively suppressing proteasome function in tumor tissues and limiting tumor growth in vivo.

CdPT inhibits tumor growth and the proteasome function in vivo. Nude mice bearing A549 xenograft tumors were treated with vehicle (Veh) or CdPT (2.5 mg kg−1 day−1, intraperitoneally) for 15 days after inoculation of cultured tumor cells. Tumor growth curves were recorded every other day. Tumor size (a), tumor images and tumor weight (b), and body weight (c) are shown. Mean ± SD. (n = 6). *P < 0.05. d Representative micrographs of immunohistochemistry staining for total ubiquitinated proteins (Ub-prs.), K48-linked ubiquitinated proteins (K48-), p21 and cleaved caspase 3 proteins in nude mouse tumor tissues. The analyses were repeated in three mouse tumor tissues and the representative images are shown
The effect of CdPT on cancer cells from leukemia patients
To explore further the translational potential of CdPT for treating human malignancy, we performed ex vivo tests for the antineoplastic effect of CdPT on the mononuclear cells from six human patients with acute myeloid leukemia (AML). Six healthy volunteers were recruited for collection of peripheral blood mononuclear cells as controls. As presented in Fig. 7a, CdPT treatment significantly reduced the viability of mononuclear cells from AML patients with an average IC50 value of 0.81 μM (0.39–1.23 μM), compared with that of CdPT-treated normal control mononuclear cells which showed an average IC50 value of 2.12 μM (1.77–2.42 μM). Treatment with 1.25, 2.5 and 5 μM CdPT obviously increased the level of total and K48-linked ubiquitinated proteins in the cancer cells from AML patients, but 1.25 and 2.5 μM CdPT had minimal effects on the mononuclear cells from normal individuals (Fig. 7b). In addition, CdPT (1.25, 2.5 and 5 μM) treatment for 24 h resulted in significant apoptosis in the mononuclear cells from patients as detected with Annexin V/PI staining by flow cytometry (Fig. 7c). These ex vivo experiments using primary human cells provide compelling evidence that CdPT was effectively inhibit proteasome function and induce cytotoxicity in the AML cancer cells, delineating the feasibility for its clinical translation.

CdPT induces cytotoxicity in cancer cells from leukemia patients. a Peripheral mononuclear cells from six leukemia patients (Patient) or six healthy volunteers (Normal) were treated with increasing doses of CdPT for 24 h, and the cell viability was detected by the MTS assay. The scatter plot of the IC50 values in each group is shown. Mean ± SD. (n = 6). *P < 0.05. b Peripheral mononuclear cells from leukemia patients (Patient) or healthy volunteers (Normal) were treated with the indicated doses of CdPT or 50 nM Velcade (Vel) for 12 h, followed by detecting total (Ub-prs.) and K48-linked (K48-) ubiquitinated proteins. GAPDH was used as a loading control. c Cancer cells from patients were treated with increasing doses of CdPT for 24 h. Typical cell apoptosis was shown. Quantified analysis is given right. Mean ± SD. (n = 3). *P < 0.05 versus control
Discussion
Metals have a number of characteristics, such as the ability to modulate the redoxstate, to be in variable coordination modes, and to exert reactivity towards organic substrates (Frezza et al. 2010), rendering them amenable to medicinal use. Metals and their salts are proved to have many medicinal applications, such as antibacterial, anti-inflammatory and anti-tumor. The discovery of cisplatin was a milestone which triggered the interest in metal complexes as potential anticancer drugs (Thompson and Orvig 2006). Although cisplatin has benefited the clinical management of several types of solid tumors, its clinical use has been drastically limited by drug resistance and severe adverse side effects, such as neuro-, hepato- and nephrotoxicity (Bruijnincx and Sadler 2008). Great efforts have been focused on the development of novel non-platinum metal based drugs with no or lower unwanted toxicity but good anticancer activity (Leon et al. 2016; Ott and Gust 2007). Cadmium (Cd) is a heavy metal pollutant widely found in the environment and is considered a human carcinogen. However, it has been reported that effective ligands, such as Disulfiram, are capable of transforming this carcinogen to potentially an efficacious anticancer drug (Li et al. 2008; Zhang et al. 2013). Pyrithione (PT) has excellent ability to chelate metals, and the PT complex with copper or zinc was previously reported to exert potent anti-tumor activity (Liu et al. 2014b; Tailler et al. 2012). In this work, we demonstrate that CdPT, a cadmium complex of pyrithione, has a better anti-tumor activity than cisplatin in A549, A549/DDP, K562 and U266 cells (Fig. 1). We have also found that CdPT induces disruption of mitochondrial membrane potential and caspase 3 activation, suggesting that the intrinsic mitochondrial pathway is involved in the CdPT-induced apoptosis (Fig. 2). In addition, our ex vivo testing showed that the IC50 values of CdPT were much lower in primary human AML mononuclear cells than that toward the normal mononuclear cells from healthy volunteers (Fig. 7). These results together provide compelling evidence that CdPT, a metal-containing agent, has a great potential for cancer therapy.
The anti-tumor activity of cisplatin, a DNA-damaging drug, is primarily thought to result from its capacity to form DNA adducts which impair cellular processes such as DNA replication and transcription that are essential to cell division and survival (Jamieson and Lippard 1999). We have previously reported that CdPT is capable of inducing phosphorylation of DNA damage response-related proteins, such as γ-H2AX, p-ATM and p-chk2 in cultured cancer cells (Zhao et al. 2016b). Thus, CdPT can act as a DNA-damage inducer. Interestingly, here we unraveled that CdPT treatment was effective in killing not only cisplatin-sensitive cancer cells (A549) but also lung cancer cells (A549/DDP) that are cisplatin-resistant (Fig. 1), indicating that CdPT may differ from cisplatin in their anti-tumor mechanisms. It has been shown that heavy metal complex could also react with important functional molecule example enzymes (Bruijnincx and Sadler 2008).
Targeting the UPS with metal-based compounds represents an emerging strategy for developing new therapeutics (Verani 2012). It has been found that metal ligands, such as Disulfiram, Clioquinol and pyrrolidine dithiocarbamate, when complexing with copper, could yield potent tumor-specific cytotoxicity via inhibiting the 20S proteasome in tumor cells (Chen et al. 2006; Daniel et al. 2005). Auranofin is a gold (I)-containing compound that has been in clinical use for rheumatoid arthritis treatment since 1985. Recently, Auranofin has attracted a great deal of attention because of its potential therapeutic application in a number of other diseases including cancer, neurodegenerative disorders, HIV, and bacterial infections (Roder and Thomson 2015). We have recently found that Auranofin is equally effective as Bortezomib in terms of proteasome inhibition properties but mechanistically it differs from Bortezomib as it inhibits 19S proteasome-associated DUBs (USP14 and UCHL5) but not the 20S proteasome (Liu et al. 2014a). Our previous studies have shown that copper pyrithione (CuPT) blocks UPS-mediated protein degradation through its potent inhibitory actions on both 19S proteasomal DUBs and 20S proteasome CT-like activities (Liu et al. 2014b). In this current work, we showed that CdPT treatment led to striking accumulation of ubiquitin-conjugated proteins in cultured cancer cell lines and primary leukemia cells. To determine the target of CdPT, we first determined the effect of CdPT on 20S proteasomes, and found that CdPT inhibited all the three proteasomal peptidases but with the IC50 higher than 5 μM, consistent with other reports showing that cadmium complex is capable of inhibiting proteasome CT-like activity (Li et al. 2008; Zhang et al. 2013). However, CdPT showed obvious cytotoxicity at a concentration much lower than the 5 μM concentration in cultured cancer cell lines. Thus, inhibition of the 20S proteasome may not be the primary mechanism underlying to the cytotoxic effects of CdPT on cancer cells.
The human genome encodes ~ 100 DUBs, which can be grouped into five distinct families (D’Arcy et al. 2011). Using the DUB enzymatic assay, we have observed that CdPT at concentrations of 0.5, 1.0 and 2 μM, completely inhibited the DUB activities of purified 26S proteasomes in vitro and partially inhibited the DUB activities in the total cell extract. This proteasomal DUB inhibition has been further verified with the Ub chain disassembly assay and the active-site-specific labeling experiment (Fig. 4). Moreover, the computational molecular docking data support that CdPT has the potential to interact with both USP14 and UCHL5 (Fig. 5a). These results demonstrate that CdPT acts more likely as a potent inhibitor of the proteasomal DUBs, USP14 and UCHL5.
USP14 and UCHL5 are members of the cysteine-containing DUB family and theoretically, the Cd atom of CdPT can potentially react with SH-containing enzymes. We have unraveled that 0.63 μM Cd2+ alone did not yield a discernible effect on the DUB activity of the proteasome but PT or CdPT at 0.63 μM potently inhibited proteasomal DUB activity (Fig. 5b). This is consistent with our previous report that PT is indispensable for NiPT to induce DUB inhibition (Zhao et al. 2016a). Interestingly, 1.25 and 2.5 μM Cd2+ showed weak inhibition on the proteasomal DUB activity which differs from Ni2+, the latter has no noticeable effect on the proteasomal DUB activity (Zhao et al. 2016a). EDTA, which is a powerful metal chelating agent, could chelate Cd2+ of CdPT and blocked CdPT from inducing cell death and from accumulating ubiquitinated proteins (Fig. 5c, d).
In addition, we also tested the in vivo anticancer activity of CdPT. Our results suggest that CdPT treatment is capable of suppressing the growth of lung cancer xenografts. Consistent with the effect of CdPT on the UPS in cultures cancer cells, we also observed increases in the levels of ubiquitinated proteins and other substratse of the UPS (e.g., p21 protein) in the tumors of CdPT-treated mice. As seen in the in vitro studies, this was accompanied by activation of caspase 3 (apoptosis) in tumors from the CdPT-treated group (Fig. 6d). The heavy metal cadmium has been reported to accumulate in kidney cells and induce oxidative stress damage (Il’yasova and Schwartz 2005; Nair et al. 2015). the body weight of mice subject to CdPT treatment remained relatively stable but that of vehicle-treated mice was gradually increased (Fig. 6c), indicating a moderate side effect of CdPT. It is possible that CdPT has some nephrotoxicity because we have previously found that CdPT at high dose induced DNA damage response in mouse kidneys (Zhao et al. 2016b).
Collectively, our findings suggest that CdPT can exert significant anticancer activity both in vitro and in vivo and that further exploration of CdPT as a therapeutic agent for cancer is warranted.
References
Adams J (2003) The proteasome: structure, function, and role in the cell. Cancer Treat Rev 29:3–9
Adams J (2004) The development of proteasome inhibitors as anticancer drugs. Cancer Cell 5:417–421
Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S, Madriaga A, Soriano F, Menon MK, Wu ZY, Kampmann M, Chen Y, Weissman JS, Aftab BT, Yakes FM, Shawver L, Zhou HJ, Wustrow D, Rolfe M (2015) Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell 28:653–665
Bruijnincx PC, Sadler PJ (2008) New trends for metal complexes with anticancer activity. Curr Opin Chem Biol 12:197–206
Chen D, Cui QC, Yang H, Dou QP (2006) Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res 66:10425–11433
Chen X, Shi X, Zhao C, Li X, Lan X, Liu S, Huang H, Liu N, Liao S, Zang D, Song W, Liu Q, Carter BZ, Dou QP, Wang X, Liu J (2014) Anti-rheumatic agent auranofin induced apoptosis in chronic myeloid leukemia cells resistant to imatinib through both Bcr/Abl-dependent and -independent mechanisms. Oncotarget 5:9118–9132
Ciechanover A (1994) The ubiquitin-proteasome proteolytic pathway. Cell 9:13–21
Coux O, Tanaka K, Goldberg AL (1996) Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem 65:801–847
Cvek B, Milacic V, Taraba J, Dou QP (2008) Ni(II), Cu(II), and Zn(II) diethyldithiocarbamate complexes show various activities against the proteasome in breast cancer cells. J Med Chem 51:6256–6258
Daniel KG, Chen D, Orlu S, Cui QC, Miller FR, Dou QP (2005) Clioquinol and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors and apoptosis inducers in human breast cancer cells. Breast Cancer Res 7:R897–908
D’Arcy P, Linder S (2012) Proteasome deubiquitinases as novel targets for cancer therapy. Int J Biochem Cell Biol 44:1729–1738
D’Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, Perego P, Sadeghi B, Hassan M, Larsson R, Linder S (2011) Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med 17:1636–1640
Dou QP, Li B (1999) Proteasome inhibitors as potential novel anticancer agents. Drug Resist Updates 2:215–223
Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C (2012) Deubiquitinases in cancer: new functions and therapeutic options. Oncogene 31:2373–2388
Frezza M, Hindo S, Chen D, Davenport A, Schmitt S, Tomco D, Dou QP (2010) Novel metals and metal complexes as platforms for cancer therapy. Curr Pharm Des 16:1813–1825
Ho YK, Bargagna-Mohan P, Wehenkel M, Mohan R, Kim KB (2007) LMP2-specific inhibitors: chemical genetic tools for proteasome biology. Chem Biol 14:419–430
Huang H, Zhang X, Li S, Liu N, Lian W, McDowell E, Zhou P, Zhao C, Guo H, Zhang C, Yang C, Wen G, Dong X, Lu L, Ma N, Dong W, Dou QP, Wang X, Liu J (2010) Physiological levels of ATP negatively regulate proteasome function. Cell Res 20:1372–1385
Il’yasova D, Schwartz GG (2005) Cadmium and renal cancer. Toxicol Appl Pharmacol 207:179–186
Jamieson ER, Lippard SJ (1999) Structure, recognition, and processing of cisplatin-DNA adducts. Chem Rev 99:2467–2498
Kazi A, Ozcan S, Tecleab A, Sun Y, Lawrence HR, Sebti SM (2014) Discovery of PI-1840, a novel noncovalent and rapidly reversible proteasome inhibitor with anti-tumor activity. J Biol Chem 289:11906–11915
Kisselev AF, Callard A, Goldberg AL (2006) Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J Biol Chem 281:8582–8590
Koulich E, Li X, DeMartino GN (2008) Relative structural and functional roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Mol Biol Cell 19:1072–1082
Leon IE, Cadavid-Vargas JF, Di Virgilio AL, Etcheverry S (2016) Vanadium, ruthenium and copper compounds: a new class of non-platinum metallodrugs with anticancer activity. Curr Med Chem 24:112–148
Li L, Yang H, Chen D, Cui C, Dou QP (2008) Disulfiram promotes the conversion of carcinogenic cadmium to a proteasome inhibitor with pro-apoptotic activity in human cancer cells. Toxicol Appl Pharmacol 229:206–214
Liu N, Li X, Huang H, Zhao C, Liao S, Yang C, Liu S, Song W, Lu X, Lan X, Chen X, Yi S, Xu L, Jiang L, Zhao C, Dong X, Zhou P, Li S, Wang S, Shi X, Dou PQ, Wang X, Liu J (2014a) Clinically used antirheumatic agent auranofin is a proteasomal deubiquitinase inhibitor and inhibits tumor growth. Oncotarget 5:5453–5471
Liu N, Liu C, Li X, Liao S, Song W, Yang C, Zhao C, Huang H, Guan L, Zhang P, Liu S, Hua X, Chen X, Zhou P, Lan X, Yi S, Wang S, Wang X, Dou QP, Liu J (2014b) A novel proteasome inhibitor suppresses tumor growth via targeting both 19S proteasome deubiquitinases and 20S proteolytic peptidases. Sci Rep 4:5240
Liu N, Huang H, Dou QP, Liu J (2015) Inhibition of 19S proteasome-associated deubiquitinases by metal-containing compounds. Oncoscience 2:457–466
Marchetti P, Castedo M, Susin SA, Zamzami N, Hirsch T, Macho A, Haeffner A, Hirsch F, Geuskens M, Kroemer G (1996) Mitochondrial permeability transition is a central coordinating event of apoptosis. J Exp Med 184:1155–1160
Mollah S, Wertz IE, Phung Q, Arnott D, Dixit VM, Lill JR (2007) Targeted mass spectrometric strategy for global mapping of ubiquitination on proteins. Rapid Commun Mass Spectrom 21:3357–3364
Nair AR, Lee WK, Smeets K, Swennen Q, Sanchez A, Thevenod F, Cuypers A (2015) Glutathione and mitochondria determine acute defense responses and adaptive processes in cadmium-induced oxidative stress and toxicity of the kidney. Arch Toxicol 89:2273–2289
Ott I, Gust R (2007) Non platinum metal complexes as anti-cancer drugs. Arch Pharm 340:117–126
Paramore A, Frantz S (2005) Bortezomib. Nat Rev Drug Discov 2:611–612
Roder C, Thomson MJ (2015) Auranofin: repurposing an old drug for a golden new age. Drugs R D 15:13–20
Shi X, Chen X, Li X, Lan X, Zhao C, Liu S, Huang H, Liu N, Liao S, Song W, Zhou P, Wang S, Xu L, Wang X, Dou QP, Liu J (2014) Gambogic acid induces apoptosis in imatinib-resistant chronic myeloid leukemia cells via inducing proteasome inhibition and caspase-dependent Bcr-Abl downregulation. Clin Cancer Res 20:151–163
Skrott Z, Cvek B (2012) Diethyldithiocarbamate complex with copper: the mechanism of action in cancer cells. Mini Rev Med Chem 12:1184–1192
Tailler M, Senovilla L, Lainey E, Thepot S, Metivier D, Sebert M, Baud V, Billot K, Fenaux P, Galluzzi L, Boehrer S, Kroemer G, Kepp O (2012) Antineoplastic activity of ouabain and pyrithione zinc in acute myeloid leukemia. Oncogene 31:3536–3546
Thompson KH, Orvig C (2006) Metal complexes in medicinal chemistry: new vistas and challenges in drug design. Dalton Trans 6:761–764
Verani CN (2012) Metal complexes as inhibitors of the 26S proteasome in tumor cells. J Inorg Biochem 106:59–67
Verma R, Aravind L, Oania R, McDonald WH, Yates JR III, Koonin EV, Deshaies RJ (2002) Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 298:611–615
Wei R, Liu X, Yu W, Yang T, Cai W, Liu J, Huang X, Xu GT, Zhao S, Yang J, Liu S (2015) Deubiquitinases in cancer. Oncotarget 6:12872–12889
Wu WK, Cho CH, Lee CW, Wu K, Fan D, Yu J, Sung JJ (2010) Proteasome inhibition: a new therapeutic strategy to cancer treatment. Cancer Lett 293:15–22
Zhang Z, Bi C, Buac D, Fan Y, Zhang X, Zuo J, Zhang P, Zhang N, Dong L, Dou QP (2013) Organic cadmium complexes as proteasome inhibitors and apoptosis inducers in human breast cancer cells. J Inorg Biochem 123:1–10
Zhao C, Chen X, Zang D, Lan X, Liao S, Yang C, Zhang P, Wu J, Li X, Liu N, Liao Y, Huang H, Shi X, Jiang L, Liu X, He Z, Dou QP, Wang X, Liu J (2016a) A novel nickel complex works as a proteasomal deubiquitinase inhibitor for cancer therapy. Oncogene 35:5916–5927
Zhao C, Chen X, Zang D, Lan X, Liao S, Yang C, Zhang P, Wu J, Li X, Liu N, Liao Y, Huang H, Shi X, Jiang L, Liu X, He Z, Wang X, Liu J (2016b) Platinum-containing compound platinum pyrithione is stronger and safer than cisplatin in cancer therapy. Biochem Pharmacol 116:22–38
Acknowledgements
This work was supported by the National High Technology Research and Development Program of China (2006AA02Z4B5), NSFC (81472762/H1609), MOE (20134423110002), Central Financial Grant of China (B16056001) (to J.L.), by Foundation for Young Innovative Talents of Guangdong Province (2016KQNCX136) and Guangdong Province Medical Science Research Foundation (A2017308) (to X.C.) and by US NIH R01 grants HL072166 and HL085629 (to X.W.).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Chen, X., Wu, J., Yang, Q. et al. Cadmium pyrithione suppresses tumor growth in vitro and in vivo through inhibition of proteasomal deubiquitinase. Biometals 31, 29–43 (2018). https://doi.org/10.1007/s10534-017-0062-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-017-0062-6