
Abstract
Based on the central role of the ubiquitin–proteasome system (UPS) in the degradation of cellular proteins, proteasome inhibition has been considered an attractive approach for anticancer therapy. Deubiquitinases (DUBs) remove ubiquitin conjugates from diverse substrates; therefore, they are essential regulators of the UPS. DUB inhibitors, especially the inhibitors of proteasomal DUBs are becoming a research hotspot in targeted cancer therapy. Previous studies have shown that metal complexes, such as copper and zinc complexes, can induce cancer cell apoptosis through inhibiting UPS function. Moreover, we have found that copper pyrithione inhibits both 19S proteasome-associated DUBs and 20S proteasome activity with a mechanism distinct from that of the classical 20S proteasome inhibitor bortezomib. In the present study, we reveal that (i) nickel pyrithione complex (NiPT) potently inhibits the UPS via targeting the 19S proteasome-associated DUBs (UCHL5 and USP14), without effecting on the 20S proteasome; (ii) NiPT selectively induces proteasome inhibition and apoptosis in cultured tumor cells and cancer cells from acute myeloid leukemia human patients; and (iii) NiPT inhibits proteasome function and tumor growth in nude mice. This study, for the first time, uncovers a nickel complex as an effective inhibitor of the 19S proteasomal DUBs and suggests a potentially new strategy for cancer treatment.
Similar content being viewed by others


Introduction
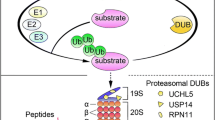
The ubiquitin–proteasome system (UPS) targets most cellular proteins for degradation and thereby is an important regulator of cell growth and apoptosis. Cancer cells, characterized by uncontrolled mitosis, require increased synthesis of growth-promoting proteins. To maintain a balance, an elevated protein-degradation capacity is apparently needed. Furthermore, compared with normal cells, cancer cells show higher sensitivity toward proteasome inhibition.1 Thus the UPS has become a promising target for anticancer strategies.1, 2, 3 The 26S proteasome is composed of two major particles: the 20S core particle and the 19S regulatory particle. The first 20S proteasome inhibitor bortezomib (Velcade) was approved by US Food and Drug Administration for the treatment of multiple myeloma in 2003.4 Many patients have benefited from the bortezomib-based therapy, and the overall survival rate of multiple myeloma has been significantly increased in the past decade. However, there are several shortcomings associated with the use of bortezomib, including relapses or resistance in some patients, severe toxic side effects and minimal effects for the treatment of solid tumors.5, 6 Therefore, there is an urgent need to discover new potent and selective proteasome inhibitor for cancer treatment.
Deubiquitinases (DUBs) are proteases that deconjugate ubiquitin from targeted proteins. The activity of these enzymes regulates the turnover rate, localization and activation of multiple proteins, which is fundamental for protein stability, cell homeostasis and multiple signaling pathways.7 Mutations in several DUBs have been linked to human diseases, including cancer and neurological disorders.8, 9, 10 In humans, three DUBs are associated with the 19S proteasome: POH1/RPN11, USP14/Ubp6, and UCHL5/Uch37. The physiological roles of the 19S DUBs are not completely understood. It is generally believed that RPN11 performs ubiquitin chain amputation by cleaving the entire ubiquitin chain from the substrate in a process coupled to degradation.11, 12 In contrast, the two cysteine-containing DUBs USP14 and UCHL5 trim ubiquitin chains from the distal end in a process antagonizing proteasomal degradation.13, 14 USP14 and UCHL5 are suggested to provide a quality control function, ensuring short or non-degradable ubiquitinated substrates to be released from the proteasome.15 A number of DUBs have been classified as oncogenes or tumor suppressors because of their regulatory functions on the activity of other proteins involved in tumor development. Therefore, recent studies have focused on search for novel anticancer drugs that target DUBs, especially the proteasome-associated DUBs.7, 14
It is known that cisplatin (CDDP), a metal compound, exerts its cytotoxic effects through the formation of covalent DNA adducts.16 The consequent genotoxic stress triggers the activation of a signaling cascade, which may lead to phosphorylation of ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia and Rad3-related), followed by recruitment and activation of other signaling proteins, such as histone 2AX (H2AX), checkpoint kinase 1 (Chk1) and Chk2, inducing cell cycle arrest or apoptosis.17, 18, 19 We and others have reported that metal-containing compounds could induce cytotoxicity in cancer cells via targeting the proteasome peptidases.20, 21, 22 Pyrithione (PT) possesses excellent metal-chelating properties, and the zinc complex of PT, for example, has been found to exert significant anticancer effects.23 Previously, we have reported that copper pyrithione (CuPT) inhibits the UPS via inhibiting both 19S proteasome and 20S proteasome activities and this inhibition has an important role in CuPT-mediated cytotoxicity.21 In this current work, the effects of nickel pyrithione (NiPT) on UPS function and cancer cell viability were investigated. We provide direct evidence that NiPT inhibits the UPS via inhibiting the 19S proteasome-associated DUBs, but not the 20S proteasome peptidases, and that the inhibition of proteasome-associated DUBs is required for NiPT-mediated cytotoxicity, unveiling a new fundamental mechanism for the anticancer effects of nickel-containing compounds.
Results
NiPT induces apoptosis in cancer cells
NiPT is formed by combining one nickel with two PTs. To investigate the effect of NiPT on the growth of human cancer cells, cultured cancer cells and normal cell lines were treated with NiPT at various concentrations for 48 h, and cell viability was measured with the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay. As shown in Figures 1a and b, NiPT decreased the cell viability in a dose-dependent manner, with the IC50 (half maximal inhibitory concentration) values of 1.72 μM in K562 cells and 4.32 μM in A549 cells. NiPT also inhibits the proliferation of U266, CDDP-resistant A549-DDP and SMMC-7721cells, with the IC50 values of 3.13, 4.17 and 2.13 μM, respectively (Supplementary Figures S1a–c). Compared with CDDP, NiPT shows comparable effect against A549 and SMMC-7721 cells but much more potent antitumor effect against the K562, U266 and A549-DDP cells. We also investigated the cytotoxicity of NiPT in the normal cell lines. We found that LO2 cells and 16HBE cells were less sensitive to NiPT than cancer cells; the IC50 values were 7.15 and >20 μM, respectively. Furthermore, NiPT exhibits lower cytotoxicity than CDDP does toward the 16HBE cells (Supplementary Figures S1d and e).

NiPT induced cytotoxicity in K562 and A549 cancer cell lines. (a, b) Cytotoxic effects of NiPT on K562 and A549 cells. K562 and A549 cells were exposed to NiPT or CDDP in various concentrations for 48 h and then were subjected to MTS assay. Data from three biological repeats are presented. Mean±s.d. (n=3). (c) Induction of apoptosis in K562 cells by NiPT. K562 cells were treated with the indicated concentrations of NiPT for 24 h, and the percentage of cells undergoing apoptosis was determined with Annexin V/PI staining followed by flow cytometry (right). The extent of cell apoptosis was summarized in the associated bar graph (lower left). Mean±s.d. (n=3). *P<0.05 versus control. (d) NiPT induced downregulation of mitochondrial membrane potential in K562 cells. K562 cells were treated with 0.25, 0.5 and 1.0 μm NiPT for 24 h, and mitochondrial membrane potential were detected by rhodamine-123 staining with flow cytometry. The extent of mitochondrial membrane potential decrease was summarized in the bar graph below. Mean±s.d. (n=3). *P<0.05 versus control. (e, f) NiPT induces cleavage of PARP and caspases-3, -8 and -9 in K562 and A549 cells. K562 (e) and A549 (f) cells were treated with either various concentrations of NiPT for 24 h or the indicated concentrations of NiPT for various times (12, 24, 36 h); PARP and caspase-3, -8 and -9 cleavage were analyzed with western blotting. GAPDH was used as a loading control.
We next analyzed the dynamics of NiPT induction of cell death in K562 and A549 cells. Exposure of K562 cells to escalating concentrations of NiPT resulted in significantly increased Annexin V/propidium iodide (PI)-positive cells as detected by flow cytometry (Figure 1c), supporting that NiPT induces apoptosis in cancer cells. It is well known that mitochondria are central to the regulation of apoptosis. We found that the integrity of mitochondrial membranes was decreased in K562 cells after NiPT treatment (Figure 1d). Consistently, in K562 and A549 cells, the precursor forms of caspase-3, -8 and -9 were decreased and reciprocally the active forms of caspase-3, -8 and -9 were increased after NiPT treatment, in parallel to increases in poly ADP-ribose polymerase (PARP) cleavage (Figures 1e and f). These results indicate that NiPT triggers caspase-dependent apoptosis in cancer cells.
NiPT triggers accumulation of ubiquitinated proteins
We and others have reported that metal complexes could directly inhibit cellular proteasome activities.20, 21, 22 To assess the effect of NiPT on the UPS, endogenous proteasome substrate proteins were first measured in human K562 and A549 cancer cells. We found that NiPT treatment induced dramatic increases of total and K48-linked ubiquitinated proteins in K562 cells (Figures 2a and b), A549 cells (Figures 2c and d) and A549-DDP cells (Supplementary Figures S2a and b) in a dose- and time-dependent manner. In addition, we created clonal HEK293 cells stably expressing a degron CL1-fused green fluorescence protein (GFPu). GFPu is a proven surrogate substrate of the UPS.24, 25 We found that NiPT also accumulated GFPu and ubiquitinated proteins in GFPu-HEK293 cells (Figures 2e and f). Velcade (bortezomib) is a classical 20S proteasome inhibitor,5, 6 whereas b-AP15 is an inhibitor of 19S proteasomal DUBs.14 We found that NiPT could induce marked increases in ubiquitinated proteins (both total and K48-linked), similarly to Velcade and b-AP15; however, CDDP showed no apparent effect on the levels of ubiquitinated proteins (Figures 2a and c and Supplementary Figure S2a) or GFPu proteins (Figure 2e). Importantly, we also found that accumulation of ubiquitinated proteins, indicative of proteasome inhibition, occurred temporally earlier than PARP cleavage (Figures 2b and d and Supplementary Figure S2b), indicating that apoptosis is induced after proteasome inhibition by NiPT in K562, A549 and A549-DDP cells.

NiPT inhibits the proteasome function in cultured cells. (a–d) Accumulation of ubiquitinated proteins. K562 (a, b) and A549 (c, d) cells were treated with either various concentrations of NiPT for 12 h or the indicated concentrations of NiPT for various times (6, 12, 18, 24 h), and then total ubiquitinated proteins (Ub-prs.), K48-linked ubiquitinated proteins (K48-) and PARP proteins were detected using western blotting analyses. Velcade (50 nM) and b-AP15 (0.5 μM) were used a positive control and CDDP (5 μM) as a negative control. GAPDH was used as a loading control. C: control. (e, f) Accumulation of GFPu, a surrogate proteasome substrate created by carboxyl fusion of an enhanced green fluorescence protein with degron CL1. GFPu-HEK293 cells, a clonal HEK293 cell line stably transfected with GFPu, were treated with Velcade (50 nM), CDDP (5 μM) and b-AP15 (0.5 μM) or the indicated concentrations of NiPT for 12 h, and then ubiquitinated proteins and GFP protein were detected with western blotting (e). Fluorescent GFPu images of the GFPu-HEK293 cells treated with NiPT (2.5 μM) or Velcade (50 nM) are shown in panel (f).
NiPT inhibits 19S proteasome-associated DUBs but not 20S proteasome activities
To determine the target of NiPT, we first detected 20S proteasome peptidase activities in vitro or in live K562, A549 and A549-DDP cells. We found that NiPT, even at a dose as high as 5 μM, did not inhibit the activities of the purified 20S proteasome chymotrypsin-like peptidases, which was distinct from Velcade (Supplementary Figure S3a). In addition, the chymotrypsin-like peptidase activities were not significantly affected by NiPT treatment in K562, A549 and A549-DDP cells under tested conditions (Supplementary Figures S3b–d). These observations are consistent with the previous findings that nickel complex could not inhibit the 20S proteasome.26, 27
Then we tested the effect of NiPT on DUB activities in cell lysates and purified 26S proteasomes. DUBs are sensitive to N-ethylmaleimide (NEM), a general inhibitor of cysteine proteases. As shown in Figure 3a, NiPT (0.5, 1.0 and 2.0 μM) yielded dose-dependent moderate inhibitory effects on the DUB activities in cell lysates; however, similar to NEM, NiPT (0.5, 1.0, 2.0 and 5 μM) could completely inhibit the DUB activities of purified 26S proteasomes (Figures 3b and c). DUB active site labelling tests were further performed by using HA-Ubiquitin-Vinyl Sulfone (HA-UbVS). HA-UbVS is a potent inhibitor against, and can bind covalently to the active site of, UCHL5 and USP14 associated with the 19S proteasome. We found that NiPT (5 and 50 μM) was able to compete with HA-UbVS for binding both UCHL5 and USP14 (Figure 3d). The cleavage of tetraubiquitin chains (Ub4) mediated by the 26S proteasome DUBs was also tested to further confirm this effect. K48-linked Ub chains were cleaved in the presence of 26S proteasomes and this was efficiently blocked by NiPT (5 and 50 μM) (Figure 3e). These results suggest that NiPT targets 19S proteasome DUBs (UCHL5 and USP14).

NiPT inhibits proteasome DUB activities. (a) Effect of NiPT on Ub-AMC cleavage by DUBs in cell lysate. Cell lysate was treated with NiPT (0.5, 1.0, 2.0 μM), and then the DUB activity at different times was recorded by using the Ub-AMC substrate. The experiment was repeated three times, yielding similar results. DUB activity was expressed as fold relative fluorescence units (RFU) difference over time. (b, c) Effect of NiPT on Ub-AMC cleavage by 26S proteasomes. Purified 26S proteasomes were treated with the indicated doses of NiPT or NEM (2 mM), and then DUB activity was kinetically detected. (d) Active-site-directed labeling of proteasomal DUBs. 26S proteasomes were treated with NiPT (5 and 50 μM) and then labeled with HA-UbVS. Labeled HA was detected by western blotting. NEM (10 mM) and b-AP15 (50 μM) were used as positive controls. (e) NiPT inhibits ubiquitin chain disassembly. K48-linked ubiquitin tetramers (Ub4) was incubated with 26S proteasomes in the absence or presence of NiPT (5 and 50 μM) for 30 min, and then ubiquitins were detected by western blotting. b-AP15 (50 μM) was used a positive control.
NiPT does not induce DNA damage
CDDP exerts its cytotoxic effects through formation of covalent DNA adducts.16 Fluorescence spectroscopy and ultraviolet (UV)-visible absorption spectroscopy were applied to investigate the competitive interaction of DNA with NiPT and a fluorescence probe, ethidium bromide (EB), as shown in Figures 4a and b. The results showed that NiPT molecules cannot compete with EB to intercalate into the DNA double-helix structure. As phosphorylation of H2AX at serine 139 (γ-H2AX) has become the gold standard for the detection of DNA damage,28 we found that CDDP, but not NiPT, produced a dose-dependent increase in the levels of γ-H2AX and phosphorylation of DNA-damage signaling proteins, including ATM, Chk1 and Chk2 in K562 (Figure 4c). Consistently, treatment for 3 h with 2.5 and 5 μM NiPT did not induce phosphorylation of DNA-damage signaling proteins in A549 cells (Figure 4d). However, NiPT treatment of A549 cells (3 h) increased the levels of total and K48-linked ubiquitinated proteins (Supplementary Figure S4). These results suggest that inhibition of proteasomal DUBs rather than induction of DNA damage is responsible for induction of cell death by NiPT.

NiPT does not induce DNA damage. (a) UV absorption spectra of NiPT with varying concentrations of DNA. Concentrations: NiPT=19.3 μM; DNA=0.00, 14.2, 28.5 and 42.7 μM for curves A through D, respectively. (b) Effect of NiPT on the fluorescence spectra of EB-DNA. EB=2 μM, DNA=14 μM, curves E–G: NiPT=12, 31, 64 μM. (c, d) NiPT does not activate DNA-damage responses. K562 cells were treated with escalating concentrations of NiPT or CDDP for 6 h (c), or A549 cells were treated with 2.5 and 5 μM NiPT or CDDP for 3 h (d), and then γ-H2AX, phosphorylated forms of ATM, chk2 and chk1 proteins were analyzed with western blotting. GAPDH was used as a loading control. C: control.
PT, rather than nickel ion, inhibits DUB activities
To investigate the role of metal ions or the chelator PT in the proteasome inhibition, we used EDTA, a powerful chelating agent, to chelate Ni2+ of NiPT as shown in Figure 5a and then investigated the effect of EDTA on NiPT-induced proteasome inhibition and cell death. We found that EDTA completely prevented NiPT from inducing ubiquitinated protein accumulation or PARP cleavage (an indicator of apoptosis) in A549 cells (Figure 5b). Similarly, co-treatment of EDTA with NiPT completely blocked NiPT from inducing cell death in A549 (Figure 5c) and K562 cells (Figure 5d). However, nickel ion alone surprisingly did not inhibit DUB activity, while both the chelator PT and NiPT had the same potency in inhibiting DUB activity in a dose-dependent manner (Figure 5e). By contrast, neither the chelator EDTA nor nickel EDTA complex (Ni-EDTA) inhibited the DUB activities (Supplementary Figure S5). We then compared the effects of PT and NiPT on ubiquitinated protein accumulation in cultured cells in the presence of either 1% or 10% fetal bovine serum (FBS). We found that PT had little effect on the levels of ubiquitinated proteins (both total and K48-linked) in 10% FBS condition but induced obvious accumulation of ubiquitinated proteins in the 1% FBS condition. However, decreasing FBS from 10% to 1% did not change the NiPT-induced accumulation of ubiquitinated proteins (Figure 5f). Furthermore, we found that bovine serum albumin (BSA) reversed PT-induced DUB inhibition more significantly than it did to NiPT (Figure 5g). These experiments demonstrate that PT, rather than nickel ion, inhibits DUB activities, that PT is the major determinant of NiPT’s potency and that Ni2+ facilitates PT’s entry into the cell.

Effect of EDTA on NiPT-induced proteasome inhibition and apoptosis. (a) Potential binding between EDTA and NiPT; EDTA binds with NiPT, forming a new product. (b) A549 cells were incubated with 5 μM NiPT in the absence or presence of 100 μM EDTA for 12 h, and then ubiquitinated proteins (Ub-prs.) and PARP proteins were detected using western blotting; GAPDH was used as a loading control. (c) A549 cells were treated as in panel (a), and then cells were stained with PI and imaged under a fluorescent microscope. The phase contrast and fluorescent images were taken and merged. Scale bar=50 μm. (d) K562 cells were treated with 5 μM NiPT or/and 100 μM EDTA for 12 h, and then the percentage of cells undergoing apoptosis was determined with Annexin V/PI staining followed by flow cytometry. E: EDTA. (e) Effect of PT, NiPT or Ni2+ on Ub-AMC cleavage by 26S proteasomes. Purified 26S proteasomes were treated with the indicated doses of PT, NiPT or Ni2+; the Ub-AMC substrate was then added to the reaction. The percentage of DUB activity was estimated by monitoring the change in substrate cleavage relative to controls (representing 100% activity). (f) Effect of FBS on PT/NiPT-induced accumulation of ubiquitinated proteins. A549 cells were incubated with 5 μM PT or NiPT in media with 1% or 10% FBS for 6 h, and ubiquitinated proteins were immunodetected. (g) Effect of BSA on PT/NiPT-induced DUB inhibition. Cell lysates were treated with PT (2 μM), NiPT (2 μM) and/or BSA (2 μM), and then Ub-AMC substrate was added to the reaction. The percentage of DUB activity was estimated by monitoring the change in substrate cleavage relative to controls (representing 100% activity). Mean±s.d. (n=3). *P<0.05.
NiPT induces cytotoxicity and proteasome inhibition in cancer cells from acute myeloid leukemia (AML) patients
We next evaluated the ex vivo antineoplastic effect of NiPT on bone marrow cells obtained from six patients with AML. Peripheral blood mononuclear cells from six healthy volunteers were used as controls. As shown in Figure 6a, NiPT decreased cell viability of primary monocytes from AML patients with IC50 values ranging from 0.110 to 0.932 μM (average: 0.479 μM) while in normal controls its IC50 values were 1.813–3.522 μM (average: 2.372 μM). NiPT treatment for 12 h at doses ranging from 0.25 to 1.0 μM resulted in significant apoptosis in the monocytes from AML patients as detected with Annexin V/PI staining followed by flow cytometry (Figure 6b) or by fluorescence microscopy (Figure 6c); however, much higher doses (from 1.25 to 5 μM) were needed to cause a comparable amount of cell death in the monocytes from healthy volunteers (Figure 6e). Treatment with NiPT significantly increased the level of total and K48-linked ubiquitinated proteins and of cleaved PARP in the cancer cells from AML patients, similarly to Velcade (Figure 6d), but elicited minimal changes in the mononuclear cells from normal controls (Figure 6f). These results demonstrate that NiPT could selectively inhibit proteasome function and induce cytotoxicity in the AML cancer cells ex vivo.

NiPT induces cytotoxicity in cancer cells from AML patients. (a) Cancer cells from six AML patients (Pt) and peripheral blood mononuclear cells from six healthy volunteers (Nm) were treated with NiPT at the indicated doses for 24 h, and the cell viability was detected with the MTS assay. The scatter plot of the IC50 values in each group is shown. Mean±s.d. (n=6). **P<0.01. (b) Cancer cells from AML patients were incubated with NiPT at the indicated doses for 24 h. Cell death was analyzed with Annexin V/PI staining followed by flow cytometry, and the results are summarized (upper right). Mean±s.d. (n=3). *P<0.05, versus control. (c) AML cancer cells and (e) healthy volunteers peripheral mononuclear cells were treated with the indicated doses of NiPT, CDDP (5 μM) or Velcade (Vel, 50 nM) for 24 h, and then cells were stained with Annexin V/PI (A/P) and imaged under a fluorescent microscope. The phase contrast and fluorescent images were taken and merged. Scale bar=50 μm. Peripheral mononuclear cells of (d) AML cancer cells and (f) healthy volunteers were treated with NiPT, CDDP (5 μM) or Velcade(Vel, 50 nM) for 24 h, followed by detecting total ubiquitinated proteins (Ub-prs.), K48-linked ubiquitinated proteins (K48-) and PARP proteins with western blotting analyses. GAPDH was used as a loading control. C: control.
NiPT accumulates proteasome substrates and selectively inhibits tumor growth in vivo
We further evaluated the in vivo effect of NiPT using nude mouse K562 and A549 xenograft models. In both K562 and A549 xenograft models, mouse body weight gradually increased in the control group while it was relatively stable in the NiPT-treated group (data not shown); the dynamic tumor growth was significantly restrained in the NiPT-treated group (Figures 7a and d), and the tumor weights were significantly lesser in the NiPT treatment group compared with the control in both tumor xenograft models (Figures 7b and e). Immunohistochemistry staining revealed that the representative proteasome substrates, including total and K48-linked ubiquitinated proteins, p27, p21 and the cleavage of caspase 3 proteins, were all significantly increased in the NiPT-treated tumors (Figures 7c and f). Together, the results show that NiPT selectively inhibits tumor growth and proteasome function in vivo.

NiPT inhibits tumor growth and the proteasome of tumor xenografts in nude mice. Nude mice bearing K562 and A549 xenograft tumors were treated with vehicle (Veh) or NiPT (5 mg/kg/day, intraperitoneally) for 15 days after inoculation of cultured tumor cells. Tumor growth curves were recorded every other day. Tumor size (a, d), tumor images and tumor weight (b, e) are shown. Mean±s.d. (n=6). **P<0.01. (c, f) Representative micrographs of immunohistochemistry staining for total ubiquitinated proteins (Ub-prs.), K48-linked ubiquitinated proteins (K48-), the indicated proteasome substrate (p21 or p27) and cleaved caspase 3 proteins in nude mouse tumor tissues. The analyses were repeated in three mouse tumor tissues and the representative images are shown.
Discussion
Nickel is a metallic element that is naturally present in the earth’s crust. Because of unique physical and chemical properties, metallic nickel and its compounds are widely used in modern industry. Human exposure to nickel occurs primarily via inhalation and ingestion; exposure to nickel compounds can have adverse effects on human health. Although the accumulation of nickel in the body through chronic exposure can lead to lung fibrosis and cardiovascular and kidney diseases, the most serious concern is related to nickel’s carcinogenic activity. Based on the high risk of pulmonary tumor related to nickel exposure, the International Agency for Research on Cancer evaluated the carcinogenicity of nickel in 1990.29 On the other hand, some nickel-containing complexes are designed for cancer therapy. Previous studies have shown that nickel complexes of 1, 2-naphthoquinone thiosemicarbazone and some S-methyl-thiosemicarbazones exhibited potent cytotoxic activities against human cancer cells.30, 31 Quercetin nickel (II) complex showed significant cytotoxicity against many tumor cell lines, and the antitumor mechanism of quercetin nickel (II) complex could involve downregulation of the survivin gene and its specific interaction with DNA.32 In this work, we explored the antitumor activity and molecular targets of the nickel ion and PT-chelating product.
The development of metal complexes such as CDDP with superior anticancer activity has had an enormous impact on cancer chemotherapy. Despite the fact that CDDP has achieved significant clinical benefit for several types of solid tumors, its effectiveness has been hampered by severe toxic side effects and drug resistance that often leads to the occurrence of secondary malignancies. The achievements of CDDP have inspired great efforts to search for novel non-platinum-containing metal species with promising anticancer activity and low toxicity.33 In this report, we found that nickel complex NiPT showed a better antitumor activity than CDDP in K562, U266 and A549-DDP cells, with lower cytotoxic activity than CDDP in the normal liver LO2 cells and bronchial epithelial 16HBE cells. In addition to the exciting potency shown to the established cancer cell lines, we also observed that the IC50 values of NiPT were much lower in mononuclear cells from AML patients than that from healthy volunteers (Figure 6). In the nude mouse xenograft model, NiPT selectively inhibited tumor growth with relatively low toxic effect on body weight (Figure 7). These results demonstrate that NiPT is a promising metal-containing drug candidate with regard to cytotoxicity, providing a great potential drug candidate in future clinical cancer therapy.
It is well known that proteasome inhibition induces apoptosis. Many metals in complexes (such as copper, zinc, gold and gallium) have been reported as proteasome inhibitors.20, 34, 35 As expected, we found that NiPT induced accumulation of ubiquitinated proteins in NiPT-treated cancer cell lines (K562, A549 and A549-DDP cells), cancer cells from AML patients and tumor tissue from nude mice. Furthermore, we have observed that NiPT exposure leads to an increased level of GFPu (a surrogate substrate of the UPS) in GFPu-HEK293 cells. It is notable that the proteasome inhibition induced by NiPT precedes cancer cell apoptosis. Taken together, proteasome inhibition is responsible for NiPT-induced apoptosis. Proteasome inhibition by NiPT may subsequently induce endoplasmic reticulum stress, which accumulates Ca2+ in cytoplasm to alter mitochondrial outer membrane permeability, leading to cytochrome c and apoptosis-inducing factor release, followed by caspase activation and apoptosis.
EDTA is a potent chelator of heavy metals outside of the cell. Scanning spectrophotometry exhibits chelation of the central zinc ion from ZnPT by addition of EDTA.36 In this paper, we found that EDTA completely prevented NiPT from inducing proteasome inhibition and apoptosis via chelating the nickel ion from NiPT. To determine the contribution of Ni2+ or of PT to the observed pharmacological effects of the NiPT complex, we compared the effects of Ni2+, PT and NiPT on proteasomal DUB activities in vitro. We found that Ni2+ by itself does not inhibit DUB activity but PT and NiPT both potently inhibit DUB activity in vitro (Figure 5e), suggesting that PT but not Ni2+ has the most important role in the NiPT-induced DUB inhibition. When tested in cultured cells in the presence of 10% FBS, however, NiPT but not PT led to significant accumulation of ubiquitinated proteins, although both were able to induce accumulation of ubiquitinated proteins in cultured cells in the presence of 1% FBS (Figure 5f), suggesting that the cell entry of PT is more potently blocked by serum albumin than that of NiPT. This is confirmed by our testing the effect of BSA on the in vitro DUB inhibition effects of PT and NiPT. BSA rescued most PT-induced DUB inhibition but only slightly attenuated NiPT-induced inhibition of the DUB activity in vitro (Figure 5g). The respective contributions of Ni2+ and PT to the DUB inhibiting activity of NiPT is further corroborated by our experiments in which PT in NiPT was replaced with EDTA and the DUB-inhibiting activity of EDTA and EDTA-Ni was tested (Supplementary Figure S5). The results show that neither EDTA nor EDTA-Ni inhibited DUB activity. Taken together, our data provide compelling support that PT is required for NiPT-induced DUB inhibition and that the presence of Ni2+ in NiPT complex remarkably reduces the binding of NiPT by serum albumin and thereby promotes the entry of NiPT into the cell where NiPT induces typical proteasome inhibition.
CDDP is primarily considered as a DNA-damaging anticancer drug, forming different types of bifunctional adducts with cellular DNA.16 Here we found that NiPT treatment was equally effective against both the CDDP-resistant A549 cell lines and the parental A549 cell lines (Figure 1b and Supplementary Figure S1b). We also found that NiPT could not directly interact with DNA. CDDP, but not NiPT, induces phosphorylation of DNA-damage signaling proteins, including ATM, H2AX, Chk1 and Chk2, in a dose-dependent manner (Figure 4). These results show that NiPT is not a DNA-damage inducer. It is well known that heavy metal could also target proteins or enzymes. Thus there must be some other targets beyond DNA involved in NiPT induction of apoptosis. DUBs, especially proteasomal DUBs, are emerging as attractive drug targets for cancer therapies. Although inhibitors of proteasomal DUBs were recently reported and shown experimentally to exhibit anticancer effects,37 their suitability for clinical use remains unknown. Metal-based compounds have been investigated for their potential as proteasome inhibitors in cancer therapy. In this paper, NiPT has the potential to inhibit 19S proteasome DUB activities (UCHL5 and USP14). This has been confirmed by the Ub chain disassembly test, active-site-directed labeling experiment and DUB activity assay with NiPT treatment at therapeutic doses. The DUB assay system used in this study is mainly suitable for the UCH and the USP families of DUBs. The enzymatic study confirmed that NiPT at 5 μM, similar to NEM, could almost completely inhibit proteasome UCHL5 and USP14 DUB activities (Figure 3). These results demonstrate that NiPT targets proteasome UCHL5 and USP14. Whether NiPT could inhibit human POH1 DUB activity needs to be investigated in the future.
Our previous studies have shown that CuPT, the chelating product of copper ion and PT, inhibits the UPS via targeting both 19S proteasome-specific DUBs and 20S proteasome peptidases. Here we found that NiPT could also inhibit 19S proteasome-associated DUBs but not the 20S proteasome. Besides targeting proteasome DUBs, both NiPT and CuPT could inhibit some other non-proteasome DUBs in the cytoplasm as well (Figure 3a and Liu et al.21), which possibly contributes to some non-proteasome-related cytotoxicity. When compared the ex vivo cytotoxic effects of CuPT and NiPT on bone marrow cells obtained from patients with AML (Pt group) and peripheral blood mononuclear cells from healthy volunteers (Nm group), NiPT and CuPT decreased cell viability of the ‘Pt group’ with an average IC50 value of 48 and 57 nM, respectively, while in the ‘Nm group’ the average IC50 values were estimated at 237 and 101 nM, respectively (Figure 6a and Liu et al.21) In conclusion, NiPT is more selective to 19S proteasome-DUBs and has lower toxicity and higher efficiency than CuPT. Future studies need to be performed to compare effects of other metals on 20S proteasomes and 19S proteasome DUBs.
Materials and methods
Materials
NiPT was synthesized in our laboratory. Other agents used include Velcade (BD Biosciences, San Jose, CA, USA); NEM, CDDP (Sigma-Aldrich Inc., St Louis, MO, USA); the Proteasome-Glo Chymotrypsin-like Cell-Based Assay Kit (Promega Bioscience, Madison, WI, USA); and b-AP15, Suc-Leu-Leu-Val-Tyr-aminomethylcoumarin (Suc-LLVY-AMC), 20S and 26S human proteasome preparations, HA-Ubiquitin-Vinyl Sulfone (HA-Ub-VS), K48-linked tetra-ubiquitin Ubiquitin-AMC (U550) (BostonBiochem, Cambridge, MA, USA). Antibodies used in this study and their sources are: anti-ubiquitin (P4D1), p27 (F-8), anti-GFP (B-2) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-p21 Waf1/Cip1 (DCS60), anti-caspase-3 (8G10), anti-caspase-8 (1C12), anti-caspase-9 (C9), anti-PARP, anti-cleaved caspase-8 (18C8), anti-K48-linkage specific polyubiquitin (D9D5), anti-phospho-histone H2A.X (Ser139) (20E3), anti-phospho-ATM (Ser1981) (D6H9), anti-phospho-Chk1 (Ser345) (133D3), anti-phospho-Chk2 (Thr68) (C13C1) (Cell Signaling Technology, Beverly, MA, USA); and anti-cleaved caspase-3, anti-cleaved caspase-9, anti-GAPDH (anti-glyceraldehyde 3-phosphate dehydrogenase), anti-HA-tag (Bioworld Technology, Inc., St Louis Park, MN, USA). PI and Annexin V-fluoroisothiocyanate (FITC)/PI apoptosis detection kit were purchased from Keygen Company (Nanjing, China). 4,6-Diamidino-2-phenylindole was from Invitrogen (Guangzhou, China). Enhanced chemiluminescence (ECL) reagents were purchased from Santa Cruz Biotechnology. The cell lines were purchased from ATCC (Manassas, VA, USA) and has been recently authenticated.
Cell viability assay
MTS assay was performed using the CellTiter 96 Aqueous One Solution reagent (Promega, Shanghai, China) to test cell viability as previously reported.38 Briefly, 10 000 cells were plated in each well and treated with either vehicle or NiPT and other agents for 48 h. Three hours before the cell culture termination, 20 μl of MTS assay reagent was added to each well of the 96-well plate. The absorbance density at wavelength 490 nm was read on a plate reader (Varioskan Flash 3001, Thermo, Waltham, MA, USA). IC50 values were calculated.
Cell death assay
Apoptosis was determined via flow cytometry following Annexin V-FITC and PI double staining.39 Cultured cells were collected and washed with the binding buffer (Sigma-Aldrich) and then incubated in the working solution (100 μl binding buffer with 0.3 μl Annexin V-FITC) for 15 min in dark; the cells were then washed and resuspended with the binding buffer. PI was added immediately before flow cytometric analysis. In addition, Annexin V/PI staining was also performed as described but in situ. The double stained cells were also imaged with an inverted fluorescence microscope equipped with a digital camera (Axio Obsever Z1, Zeiss, Jena, Germany).
To monitor temporal changes in the incidence of cell death in the live culture condition, PI was added to the cell culture medium, and at the desired sequential time points, the cells in the culture dish were imaged with an inverted fluorescence microscope.40
Mitochondrial membrane potential
The mitochondrial membrane potential (ΔΨm) of cancer cells was assayed using rhodamine-123 (Sigma-Aldrich) staining as previously reported.41 Cells treated with various concentrations of NiPT or controls were stained with 1 μM of rhodamine-123 for 1 h at 37 °C. Following the staining, the cells were washed and harvested for flow cytometric analysis.
Western blotting analysis
Whole-cell lysates were prepared in RIPA buffer (1 × phosphate-buffered saline, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS)) supplemented with 10 mM β-glycerophosphate, 1 mM sodium orthovanadate, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1 × Roche Complete Mini Protease Inhibitor Cocktail (Roche, Indianapolis, IN, USA). SDS–PAGE (polyacrylamide gel electrophoresis), transferring and immunodetection were performed as previously described.42 In brief, an equal amount of total protein extracts were fractionated by 12% SDS–PAGE and electrically transferred onto a polyvinylidene difluoride (PVDF) membrane. Primary antibodies and appropriate horseradish peroxidase-conjugated secondary antibodies were used to detect the designated proteins. The bound secondary antibodies on the PVDF membrane were reacted to the ECL detection reagents (Santa Cruz Biotechnology) and detected by exposing to X-ray films (Kodak, Rochester, NY, USA). The cell lines were purchased from ATCC (American Type Culture Collection) and have been recently authenticated.
Sample collection from human subjects and the isolation and culture of human mononuclear cells
Peripheral blood samples of normal control individuals were obtained from Guangzhou Blood Center and peripheral bone marrow samples of AML patients were obtained from discarded material utilized for routine laboratory tests at the Department of Hematology, Guangzhou First Municipal People’s Hospital of Guangzhou Medical University. The use of these materials is approved by the Ethics Committee of these two Institutions and is with the permission of the patients and volunteers. A total of six patients with AML and six healthy volunteers were included in this preclinical study. Mononuclear cells were isolated from either peripheral blood or bone marrow samples by Ficoll-Paque (Pharmacia, Uppsala, Sweden) density gradient. The mononuclear cells were cultured in RPMI 1640 culture medium with 15% FBS.
Peptidase activity assay
Fluorogenic substrate Suc-LLVY-AMC was used to assess chymotrypsin-like activity of the 20S proteasome.43 To evaluate in vivo proteasome inhibition, cancer cells were treated with NiPT or bortezomib for 4 h. The cells were lysed in ice-cold lysis buffer. Equal amounts of protein from each sample were then incubated at 37 °C with 50 μM fluorogenic substrate. To assay for direct inhibition of the 20S proteasome in vitro, purified human 20S proteasomes were incubated with the agent to be tested for 60 min at 37 °C before the addition of the fluorogenic substrate. Fluorescence intensity was measured using a spectrophotometer at excitation of 350 nm and emission of 438 nm (Varioskan Flash 3001, Thermo).
DUB activity assay
This was performed as reported.14 Briefly, cell lysate (5 μg) or 26S proteasome (25 nM) was dissolved in ice-cold DUB buffer (50 mM Tris-HCl (pH 7.5), 250 mM sucrose, 5 mM MgCl2 and 1 mM PMSF) and pretreated with NiPT (5 μM) or NEM (2 mM) for 15 min and then incubated with Ub-AMC substrate in a 100-μl reaction volume at 25 °C. AMC released from substrate cleavage was temporally recorded with a microplate reader (Varioskan Flash 3001, Thermo).
DUB active-site-directed labeling assays
Purified 26S proteasomes (25 nM) were dissolved in DUB buffer (25 mM Tris-HCl pH 7.4, 5 mM MgCl, 20 mM NaCl, 200 μM ATP) and then treated with NiPT (5, 50 μM) for 10 min before they were incubated with HA-UbVS for 1 h at 37 °C, followed by boiling in the reducing sample buffer and fractionated via SDS–PAGE. After transferred to PVDF membranes, HA-UbVS-labeled DUBs were immunodetected using anti-HA antibodies.
Ubiquitin chain disassembly
In vitro disassembly of purified polyubiquitin chains (K48-linked) was performed as described earlier.44 Purified 26S proteasomes (25 nM) were preincubated with either vehicle or NiPT for 10 min in vitro before mixed with K48-linked polyubiquitin chains (1 mg) in the DUB reaction buffer for 30 min at 37 °C. The reaction product was then fractionated using SDS–PAGE, transferred to PVDF membrane and assessed for the extent of chain disassembly via immunodetection of ubiquitin.
Spectroscopic studies on DNA interaction
Calf thymus DNA was purchased from the Sigma Company (Steinheim, Germany). All the spectroscopic titration were carried out in the buffer containing 50 mM Tris-HCl and 20 mM NaCl (pH 7.4) at room temperature. The A260 to A280 ratio was 1.84, indicating that the DNA was sufficiently free of protein. The DNA concentration per nucleotide was determined by absorption spectroscopy using the known molar extinction coefficient value of 6600 M/cm at 260 nm. The UV spectrophotometric measurements were performed at a fixed concentration of NiPT while varying DNA concentration using a UV-Vis spectrophotometer, operating from 200 to 600 nm. The fluorescence spectrophotometric measurements were carried out with a spectrofluorometer; both excitation and emission slits were set as 10 nm. In competition binding complexes with EB-DNA, DNA and EB concentrations were 14 and 2 μM, respectively, while NiPT varied from 0 to 64 μM. The fluorescence spectra were measured in the wavelength range of 550–800 nm, with the excitation wavelength at 526 nm.
Nude mouse xenograft model
All animal protocols used were approved by the Institutional Animal Care and Use Committee of Guangzhou Medical University. The mice were obtained from Guangdong Laboratory Animal Monitoring Institute (SCXK2008-2002). The nude Balb/c mice were housed in barrier facilities with a 12-h light–dark cycle, with food and water available ad libitum. Approximately 1 × 107 of K562 or A549 cells were inoculated subcutaneously on the flanks of 5-week-old male nude mice. Seventy-two hours after inoculation, the mice were randomly assigned to two groups and treated with either vehicle (10% dimethyl sulfoxide, 30% polyethylene glycol 400 and 60% normal saline) or NiPT (5 mg/kg/day) for 15 days. Tumor volumes were recorded and calculated as we previously described.45
Immunohistochemistry
Formalin-fixed xenografts were embedded in paraffin and sectioned according to standard techniques as we previously reported.39 Tumor xenograft sections (4 μm) were immunostained using the MaxVision Kit (Maixin Biol, Fuzhou, China) according to the manufacturer’s instructions. The primary antibodies were used as indicated. In all, 50 μl MaxVision reagent was applied to each slide. Color was developed with 0.05% diaminobenzidine and 0.03% H2O2 in 50 mM Tris-HCl (pH 7.6), and the slides were counterstained with hematoxylin. A negative control for every antibody was also included for each xenograft specimen by substituting the primary antibody with preimmune rabbit serum.
Statistical analysis
All the results were expressed as mean±s.d. where applicable. GraphPad Prism 4.0 software (GraphPad Software, San Diego, CA, USA) was used for statistical analysis. Differences between two groups were evaluated for statistical significance using two-tailed Student’s t-test. For testing differences among three or more independent groups, one-way analysis of variance or, when appropriate, two-way analysis of variance, followed by the Holm–Sidak test for pair-wise comparisons, were performed. P-value of <0.05 was considered statistically significant.
References
Adams J . The proteasome: structure, function, and role in the cell. Cancer Treat Rev 2003; 29: 3–9.
Mani A, Gelmann EP . The ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol 2005; 23: 4776–4789.
Burger AM, Seth AK . The ubiquitin-mediated protein degradation pathway in cancer: therapeutic implications. Eur J Cancer 2004; 40: 2217–2229.
Paramore A, Frantz S . Bortezomib. Nat Rev Drug Discov 2003; 2: 611–612.
Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP . Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets 2011; 11: 239–253.
Adams J . The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004; 5: 417–421.
Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C . Deubiquitinases in cancer: new functions and therapeutic options. Oncogene 2012; 31: 2373–2388.
Singhal S, Taylor MC, Baker RT . Deubiquitylating enzymes and disease. BMC Biochem 2008; 9: S3.
Sacco JJ, Coulson JM, Clague MJ, Urbe S . Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB Life 2010; 62: 140–157.
Todi SV, Paulson HL . Balancing act: deubiquitinating enzymes in the nervous system. Trends Neurosci 2011; 34: 370–382.
Verma R, Aravind L, Oania R, McDonald WH, Yates JR 3rd, Koonin EV et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002; 298: 611–615.
Koulich E, Li X, DeMartino GN . Relative structural and functional roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Mol Biol Cell 2008; 19: 1072–1082.
Yao T, Song L, Xu W, DeMartino GN, Florens L, Swanson SK et al. Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat Cell Biol 2006; 8: 994–1002.
D'Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med 2011; 17: 1636–1640.
Ventii KH, Wilkinson KD . Protein partners of deubiquitinating enzymes. Biochem J 2008; 414: 161–175.
Jamieson ER, Lippard SJ . Structure, recognition, and processing of cisplatin-DNA adducts. Chem Rev 1999; 99: 2467–2498.
Pabla N, Huang S, Mi QS, Daniel R, Dong Z . ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J Biol Chem 2008; 283: 6572–6583.
Roos WP, Kaina B . DNA damage-induced cell death by apoptosis. Trends Mol Med 2006; 12: 440–450.
Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM . A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000; 10: 886–895.
Milacic V, Chen D, Giovagnini L, Diez A, Fregona D, Dou QP . Pyrrolidine dithiocarbamate-zinc(II) and -copper(II) complexes induce apoptosis in tumor cells by inhibiting the proteasomal activity. Toxicol Appl Pharmacol 2008; 231: 24–33.
Liu N, Liu C, Li X, Liao S, Song W, Yang C et al. A novel proteasome inhibitor suppresses tumor growth via targeting both 19S proteasome deubiquitinases and 20S proteolytic peptidases. Sci Rep 2014; 4: 5240.
Verani CN . Metal complexes as inhibitors of the 26S proteasome in tumor cells. J Inorg Biochem 2012; 106: 59–67.
Tailler M, Senovilla L, Lainey E, Thepot S, Metivier D, Sebert M et al. Antineoplastic activity of ouabain and pyrithione zinc in acute myeloid leukemia. Oncogene 2012; 31: 3536–3546.
Bence NF, Sampat RM, Kopito RR . Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001; 292: 1552–1555.
Liu J, Chen Q, Huang W, Horak KM, Zheng H, Mestril R et al. Impairment of the ubiquitin-proteasome system in desminopathy mouse hearts. FASEB J 2006; 20: 362–364.
Cvek B, Milacic V, Taraba J, Dou QP . Ni(II), Cu(II), and Zn(II) diethyldithiocarbamate complexes show various activities against the proteasome in breast cancer cells. J Med Chem 2008; 51: 6256–6258.
Frezza M, Hindo SS, Tomco D, Allard MM, Cui QC, Heeg MJ et al. Comparative activities of nickel(II) and zinc(II) complexes of asymmetric [NN'O] ligands as 26S proteasome inhibitors. Inorg Chem 2009; 48: 5928–5937.
Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A . H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004; 3: 959–967.
Denkhaus E, Salnikow K . Nickel essentiality, toxicity, and carcinogenicity. Crit Rev Oncol Hematol 2002; 42: 35–56.
Chen J, Huang YW, Liu G, Afrasiabi Z, Sinn E, Padhye S et al. The cytotoxicity and mechanisms of 1,2-naphthoquinone thiosemicarbazone and its metal derivatives against MCF-7 human breast cancer cells. Toxicol Appl Pharmacol 2004; 197: 40–48.
Atasever B, Ulkuseven B, Bal-Demirci T, Erdem-Kuruca S, Solakoglu Z . Cytotoxic activities of new iron(III) and nickel(II) chelates of some S-methyl-thiosemicarbazones on K562 and ECV304 cells. Invest New Drugs 2010; 28: 421–432.
Tan J, Zhu L, Wang B . From GC-rich DNA binding to the repression of survivin gene for quercetin nickel (II) complex: implications for cancer therapy. Biometals 2010; 23: 1075–1084.
Florea AM, Busselberg D . Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel) 2011; 3: 1351–1371.
Chen D, Frezza M, Shakya R, Cui QC, Milacic V, Verani CN et al. Inhibition of the proteasome activity by gallium(III) complexes contributes to their anti prostate tumor effects. Cancer Res 2007; 67: 9258–9265.
Liu N, Li X, Huang H, Zhao C, Liao S, Yang C et al. Clinically used antirheumatic agent auranofin is a proteasomal deubiquitinase inhibitor and inhibits tumor growth. Oncotarget 2014; 5: 5453–5471.
Dinning AJ, Al-Adham IS, Austin P, Charlton M, Collier PJ . Pyrithione biocide interactions with bacterial phospholipid head groups. J Appl Microbiol 1998; 85: 132–140.
Tian Z, D'Arcy P, Wang X, Ray A, Tai YT, Hu Y et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014; 123: 706–716.
Li X, Liu S, Huang H, Liu N, Zhao C, Liao S et al. Gambogic acid is a tissue-specific proteasome inhibitor in vitro and in vivo. Cell Rep 2013; 3: 211–222.
Chen X, Shi X, Zhao C, Li X, Lan X, Liu S et al. Anti-rheumatic agent auranofin induced apoptosis in chronic myeloid leukemia cells resistant to imatinib through both Bcr/Abl-dependent and -independent mechanisms. Oncotarget 2014; 5: 9118–9132.
Huang H, Zhang X, Li S, Liu N, Lian W, McDowell E et al. Physiological levels of ATP negatively regulate proteasome function. Cell Res 2010; 20: 1372–1385.
Shi X, Lan X, Chen X, Zhao C, Li X, Liu S et al. Gambogic acid induces apoptosis in diffuse large B-cell lymphoma cells via inducing proteasome inhibition. Sci Rep 2015; 5: 9694.
Huang H, Liu N, Guo H, Liao S, Li X, Yang C et al. L-carnitine is an endogenous HDAC inhibitor selectively inhibiting cancer cell growth in vivo and in vitro. PLoS One 2012; 7: e49062.
Yang H, Zhou P, Huang H, Chen D, Ma N, Cui Q et al. Shikonin exerts antitumor activity via proteasome inhibition and cell death induction in vitro and in vivo. Int J Cancer 2009; 124: 2450–2459.
Dayal S, Sparks A, Jacob J, Allende-Vega N, Lane DP, Saville MK . Suppression of the deubiquitinating enzyme USP5 causes the accumulation of unanchored polyubiquitin and the activation of p53. J Biol Chem 2009; 284: 5030–5041.
Shi X, Chen X, Li X, Lan X, Zhao C, Liu S et al. Gambogic acid induces apoptosis in imatinib-resistant chronic myeloid leukemia cells via inducing proteasome inhibition and caspase-dependent Bcr-Abl downregulation. Clin Cancer Res 2014; 20: 151–163.
Acknowledgements
We thank Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-Sen Memorial Hospital and Sun Yat-Sen University for flow cytometry detection. This work was supported by the National High Technology Research and Development Program of China (2006AA02Z4B5), NSFC (81272451/H1609, 81472762/H1609), MOE (20134423110002) and Guangdong Key Laboratory of Urology (2010A060801016) (to JL) and in part by US NIH grants HL072166 and HL085629 (to XW).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
About this article

Cite this article
Zhao, C., Chen, X., Zang, D. et al. A novel nickel complex works as a proteasomal deubiquitinase inhibitor for cancer therapy. Oncogene 35, 5916–5927 (2016). https://doi.org/10.1038/onc.2016.114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.114
- Springer Nature Limited
This article is cited by
-
Deubiquitinases in hematological malignancies
Biomarker Research (2021)
-
Regulation of Bax-dependent apoptosis by mitochondrial deubiquitinase USP30
Cell Death Discovery (2021)
-
Pyrithione metal (Cu, Ni, Ru) complexes as photo-catalysts for styrene oxide production
Scientific Reports (2021)
-
The deubiquitylase UCHL3 maintains cancer stem-like properties by stabilizing the aryl hydrocarbon receptor
Signal Transduction and Targeted Therapy (2020)
-
Inhibition of EGFR signaling with Spautin-1 represents a novel therapeutics for prostate cancer
Journal of Experimental & Clinical Cancer Research (2019)