
Abstract
The toxic effect of Ni2+ on photosynthetic electron transport was studied in a photosystem II submembrane fraction. It was shown that Ni2+ strongly inhibits oxygen evolution in the millimolar range of concentration. The inhibition was insensitive to NaCl but significantly decreased in the presence of CaCl2. Maximal chlorophyll fluorescence, together with variable fluorescence, maximal quantum yield of photosystem II, and flash-induced fluorescence decays were all significantly declined by Ni2+. Further, the extrinsic polypeptides of 16 and 24 kDa associated with the oxygen-evolving complex of photosystem II were depleted following Ni2+ treatment. It was deduced that interaction of Ni2+ with these polypeptides caused a conformational change that induced their release together with Ca2+ from the oxygen-evolving complex of photosystem II with consequent inhibition of the electron transport activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Divalent metal cations are widespread pollutants that are phytotoxic. Most of them are absorbed by the plant roots where they can accumulate or translocate depending on metal concentration. Prolonged exposure may lead to important bioaccumulation and translocation towards the leaves where the photosynthetic apparatus becomes an inhibitory target (Clijsters and van Asche 1985; Carpentier 2002).
Nickel is a toxic divalent cation that is readily absorbed by plant roots and translocated to the leaf tissues where it clearly affects photosynthesis (Tripathy et al. 1981; Mohanty et al. 1989; Krupa et al. 1993). Its precise mode of action has not been studied in great detail. A significant deterioration of the chloroplast grana structure was reported in nickel-treated cabbage plants (Molas 1997). The content of photosynthetic pigment was also reduced to various extents depending on growth stage (Veeranjaneyulu and Das 1982; Sheoran et al. 1990; Krupa et al. 1993; Xyländer and Braune 1994). Along with other toxic divalent cations, this heavy metal was also suggested from Fourier transform infrared and electron spin resonance spectroscopy to dissociate membrane protein complexes thus increasing lipid-protein interaction (Szalontai et al. 1999). Peroxidation of membrane lipids due to the induction of free radical reactions by Ni2+ was proposed to account for thylakoid membrane deterioration (Molas 1997).
Contradictory results were obtained regarding its mode of action on the photosynthetic electron transport reactions. Nickel was reported to mainly inactivate photosystem I in Cylindrospermum (Singh et al. 1989) whereas PSII was proposed as the main target in isolated chloroplasts (Tripathy et al. 1981). Most in vivo and in vitro studies indicated an inhibition of electron transport on the donor side of PSII that was illustrated by a decreased Chl fluorescence yield (Tripathy et al. 1981; Mohanty et al. 1989; Singh et al. 1989). However, the inhibition in PSII was also discussed in terms of a modification of the binding site for QB, the secondary quinone acceptor of PSII (Mohanty et al. 1989; El-Sheekh 1993). The primary donor of PSII, P680, itself was also postulated as a direct target of Ni2+ action (Tripathy et al. 1981, 1983).
The most precise studies regarding the mode of action of toxic metal cations were performed in isolated thylakoid membranes or photosystem submembrane fractions where the electron transfer components are more easily accessible to the inhibitors (Carpentier 2002). In this study, we used submembrane fractions enriched in PSII to provide a further clarification of the inhibitory action of Ni2+ in this photosystem. The presence of an active site at the oxygen-evolving complex (OEC) is derived from the data obtained from Chl fluorescence induction and flash-induced decay measurements. The cause of the inhibition is discussed in terms of the release of extrinsic polypeptides and Ca2+ from the OEC.
Materials and methods
Isolation of PSII submembrane fractions
PSII submembrane fractions were isolated from spinach (Spinacea oleracea L.) according to Berthold et al. (1981) with the modification described elsewhere (Rashid et al. 1991). The PSII preparations were finally resuspended in 400 mM sucrose and 20 mM MES-NaOH (pH 6.3) and were stored at −80°C until use. When specified, the extrinsic polypeptides of 17, 24 and 33 kDa were removed by alkali-salt treatment or the polypeptides of 17 and 24 kDa were removed by 1 M NaCl treatment using the procedure described by Nakatani (1984).
Polypeptides analysis
Nickel treatments (10-min incubation) were performed in the dark at 22°C. The assay medium contained 400 mM sucrose, 20 mM MES-NaOH (pH 6.3), and nickel at the specified metal/Chl ratios. To determine the polypeptides released by nickel, the PSII preparations were harvested immediately after treatment by an 8-min centrifugation in an Eppendorf microcentrifuge (12,400 rpm, 13,600g). The pellets were washed once in 400 mM sucrose, 20 mM MES-NaOH (pH 6.3) and were used for polypeptide analysis. The first supernatants were further centrifuged (40 min) to remove remaining membrane fragments. The supernatants were then concentrated against a sucrose gradient and dialysed against the above buffer before analysis by polyacrylamide gel electrophoresis. The latter was performed at room temperature using miniature slab gels (Bio-Rad Laboratories, Hercules, California) containing 13% acrylamide and 6 M urea. The gels were stained with Coomassie brilliant blue and the polypeptide content was analysed with the Gel-Doc 2000 system (Bio-Rad Laboratories, Hercules, California).
Manganese determination
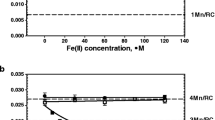
For manganese concentration measurement, PSII particles at 0.5 mg/ml were treated with NiCl2 and centrifuged at 35,000g for 30 min. Manganese concentration was then determined in the supernatant with an Analyst 100 atomic absorption spectrophotometer (Perkin Elmer, Wellesley, MA, USA) at a wavelength of 279.5 nm using an air/acetylene flame.
Oxygen evolution
Oxygen evolution was measured at 22°C using Oxylab system (Hansatech Instruments, Norfolk, England). The assay medium contained 400 mM sucrose, 20 mM MES-NaOH (pH 6.3), 0.35 mM 2,6-dichlorobenzoquinone as PSII electron acceptor, 10 μg Chl/ml and the specified concentrations of nickel. In the absence of nickel, the oxygen evolution rates were 700–800 μmol O2 / mgChl · h.
Fluorescence induction
Fluorescence induction measurements were performed at room temperature using Plant Efficiency Analyser (Hansatech, King’ Lynn, Norfolk, UK). The assay medium contained 400 mM sucrose, 20 mM MES-NaOH (pH 6.3), 25 μg Chl/ml and the specified concentrations of nickel. Dark-adapted samples were excited with saturating red actinic light (peaking at >655 nm and intensity of 4000 μmol m−2 s−1) provided by light emitting diodes. As the fluorescence signal during the first 40 μs is ascribed for artifacts due to delay in response time of the instrument, these data were not included in analyses of FI traces. For quantitative analysis, FI traces were fitted with the sum of three first-order kinetics by non-linear regression using Sigma Plot (SSI, Richmond, California, USA):
where F(t) is the fluorescence at time t, F 0 is the initial fluorescence, A O-J, A J-I and A I-P are the amplitudes, k O-J, k J-I and k I-P are the rate constants of the O-J, J-I and I-P steps of the fluorescence transient (Pospišil and Dau 2002; Boisvert et al. 2006).
Flash-induced fluorescence decay kinetics
In order to detect the reduction and oxidation kinetics of QA, Chl fluorescence rise and its relaxation in the dark were measured with a pulse amplitude double modulated fluorometer (PAM, Walz, Effeltrich, Germany) as described previously (Ono et al. 1995; Putrenko et al. 1999). The fluorescence signal was collected with the PDA 100 data acquisition system through WinControl software (Walz, Effeltrich, Germany) using a 20 μs/data point window with measuring light modulated at 100 kHz. The duration of the measuring time with the weak modulated light at a frequency of 1.6 kHz and the measuring light modulated at 100 kHz was less than 290 ms. The single turn over saturating flash generated from XST 103 (Walz, Effeltrich, Germany) flash lamp used here reaches its maximum intensity within 8 μs. This intensity of the flash completely decays within 50 μs. Since the actual fluorescence signal stabilizes 100 μs after the flash was applied, the data pertaining to this time-period was not used for the analyses of \( {\hbox{Q}}_{\hbox{A}}^ - \) oxidation kinetics. PSII submembrane fractions at a Chl concentration of 25 μg/ml were incubated for 10 min at room temperature in complete darkness without or with 0.1 or 0.2 mmol NiCl2/mg Chl before initiating the fluorescence measurements. The signal to noise ratio was improved by measuring Chl fluorescence rise and its decay in the dark from nine independent samples of the same preparation. These original traces were averaged with WinControl software to estimate the half-times and amplitudes of the fluorescence decay phases using the following three-exponential function:
where F(t) is the fluorescence value at time t, k n is the rate constant, A n is the amplitude of the fluorescence relaxation phases, and F′ is the stable minimal fluorescence at the end of decay. The fluorescence decay curves could not be fit with a bi-exponential function.
Results
The influence of Ni2+ on the oxygen-evolving activity of PSII submembrane fractions isolated from spinach is shown in Fig. 1. For direct comparison with other figures, NiCl2 concentrations are depicted as mmol/mg Chl. Because the Chl concentration in the assay medium for oxygen evolution was 10 μg per ml, 0.1 mmol NiCl2/mg Chl is equivalent to 1 mM NiCl2 in the experiment of Fig. 1. It is clear in Fig. 1 that a 10-min incubation of the PSII submembrane fractions in the presence of Ni2+ strongly inhibited oxygen evolution in the mM range of NiCl2 concentration. Identical inhibitory profiles were obtained with NiBr2 (not shown). Addition of 10 mM NaCl to the assay medium during Ni2+ treatment did not modify the inhibitory profile showing chloride ions did not affect Ni2+ action. However, 5 mM CaCl2 introduced either before or after addition of Ni2+ to the assay medium greatly reduced the inhibitory action (Fig. 1). Thus, Ca2+ but not Cl− could prevent the deleterious effect of nickel.

Inhibition of oxygen evolution activity in PSII submembrane fractions after a 10-min incubation under various concentrations of NiCl2 added alone (■), or with 10 mM NaCl (▴), with 5 mM CaCl2 introduced in the assay medium before incubation with nickel (χ), or with 5 mM CaCl2 introduced after incubation with nickel (□). For this and other figures, error bars represent the standard deviation and the data presented are the average of three independent measurements obtained form different samples
The protective effect of Ca2+ is documented in more detail in Fig. 2. The inhibition obtained with 1.5 and 3 mM Ni2+ was gradually relieved with increasing CaCl2 concentrations. In Fig. 2 (insert), the double-reciprocal plot of the data presented in Fig. 2 indicates that Ni2+ behaves as a competitive inhibitor with respect to Ca2+ with a small component of mixed inhibition.

Decreased inhibitory effect of nickel on oxygen evolution in the presence of various CaCl2 concentrations added to the assay medium prior to incubation with 1.5 mM (■) or 3 mM (▴) NiCl2. Inset: double-reciprocal plot of the data in Fig. 2
The influence of Ni2+ on the polypeptide profile of the PSII submembrane fractions was analysed using polyacrylamide gel electrophoresis (Fig. 3). In lane 4, it is shown that a 10-min incubation of the PSII submembrane fragments in the presence of 1 mmol NiCl2/mg Chl produced strong depletion of two polypeptides of apparent molecular weight of 17 and 24 kDa as compared to the control (lanes 2 and 3). These polypeptides were recovered in the supernatant of the treated samples as seen in lane 5. The polypeptides in the supernatants obtained after NaCl–Tris (pH 9.2) treatment and NaCl treatment alone are shown in lane 6 and 7, respectively. NaCl–Tris treatment removes the extrinsic polypeptides of 17, 24, and 33 kDa associated with the OEC of PSII and NaCl treatment depletes the 17 and 24 kDa polypeptides. The proteins removed by nickel treatment (lane 5) corresponded to the 17 and 24 kDa polypeptides removed by NaCl treatment. However, the 33-kDa polypeptide that is also removed by NaCl–Tris treatment remained unaffected by Ni2+ treatment.

Effect of a 10-min incubation of PSII submembrane fractions in the presence of 1 mmol NiCl2/mg Chl on the polypeptide profile. Lane 1, molecular weight standards; 2 and 3, control PSII; 4, nickel-treated PSII; 5, supernatant of nickel-treated PSII; 6, supernatant of alkali-salt-treated PSII; 7, 1 M NaCl-treated PSII. Numbers on the left side indicate apparent molecular masses (kDa) of the molecular markers
The polypeptide content for the extrinsic proteins of 17, 24, and 33 kDa following incubation of 10 min with various concentrations of Ni2+ is shown in Fig. 4. At any Ni2+ concentration, the amount of 33 kDa polypeptide in the PSII submembrane fractions remained unaltered. In contrast, the polypeptides of 17 and 24 kDa were progressively lost up to about 70% of their initial content. Ni2+ concentration that caused about 50% of the protein depletion effect was about 0.3 mmol/mg Chl for the 17 kDa protein and 0.5 mmol/mg Chl for the 24 kDa protein. This concentration range is very similar to the effect on oxygen evolution (Fig. 1). Thus, protein depletion from the OEC occurred simultaneously with inhibition of the oxygen-evolving activity. The polypeptide release also coincided with the loss of about two Mn per PSII from the OEC (result not shown).

Extrinsic polypeptide content as a function of NiCl2 concentration: (■) 17 kDa polypeptide; (▴) 24 kDa polypeptide; (•) 33 kDa polypeptide
The action of Ni2+ on variable Chl fluorescence was studied. In Fig. 5A, both the initial Chl fluorescence obtained in dark adapted samples, F 0, and the maximal Chl fluorescence measured under saturating light, F m, are shown. F 0 was not significantly modified in the presence of Ni2+. However, Fm decreased as Ni2+ concentration was raised in a manner similar to the range of concentration used for oxygen evolution measurements. The decline in Fm coincided with a decrease in F v = F m − F 0 and consequently, the maximal quantum yield of PSII measured as F v/F m also decreased (Fig. 5B). Because F 0 was barely modified by Ni2+, the inhibitory action on the quantum yield of PSII was due to the changes in Fv. Calculation of F v/F 0, a parameter that simultaneously takes into account the variations in F m and F 0 (Babani and Lichtenthaler 1996), provided similar traces as Fig. 5B (not shown).

Effect of increasing NiCl2 concentration in PSII submembrane fractions on the Chl fluorescence parameters F m, F 0, and (F m − F 0)/ F m. Here and in the following figures, a.u. is arbitrary units
To gain more information on the Chl fluorescence induction properties of Ni2+-treated PSII, we analyzed the O-J–I-P induction traces. These traces illustrate the progressive reduction of the quinones located at the acceptor side of PSII with three main phases corresponding to O-J, J-I, and I-P (Lazár 1999, 2006; Zhu et al. 2005). Such traces are shown in Fig. 6A. As expected from the decreased F v (Fig. 5B), Chl fluorescence intensity decreased as NiCl2 concentration was increased. The three phases of the induction are deconvoluted from the experimental induction traces using a sum of three successive exponential rises (Pospišil and Dau 2002; Boisvert et al. 2006) as described in Materials and methods. The three deconvoluted phases are shown in Fig. 6B. The good fitting obtained by this type of non-linear regression shows that the method can be used as an excellent approximation of fluorescence induction traces and to quantitatively estimate the contribution of each phase. Each phase has a defined amplitude and half-time that are represented in Fig. 7 for O-J and I-P as a function of Ni2+ concentration. The J-I phase is negligible in PSII submembrane fractions (Pospišil and Dau 2000). The decreasing amplitudes of O-J and I-P (Fig. 7A) were consistent with Fv variations, showing that both phases were affected similarly. However, the half-time for I-P greatly increased in proportion to the rising Ni2+ concentrations (Fig. 7B).

(A) Chl a fluorescence induction traces in PSII submembrane fractions treated with various concentrations (mmol/mg Chl) NiCl2: 1, control; 2, 0.1; 3, 0.7; 4, 1.5. (B) Control trace from Fig. 6a of the fluorescence (open circles) and its simulation (full line) by three exponential components (O-J, J-I, and I-P) added to F 0

(A) Amplitudes and (B) half-times of the O-J and I-P phases simulated by exponential components at various NiCl2 concentrations. The half-times were normalized to unity for control (control values were t ½O-J = 0.34 ms and t½I-P = 94 ms)
The fluorescence properties of PSII submembrane fractions submitted to a single turn over flash were also studied and the results are presented in Fig. 8. Typical fluorescence decay kinetics on a logarithmic scale are shown in Fig. 8A. The fluorescence rise is due to the reduction of QA, the primary quinone acceptor of PSII, and the decay insured in the dark is due to the reoxidation of \( {\hbox{Q}}_{\hbox{A}}^ - \). The dark decay was modelled with a three-exponential function. The fast component is attributed to the reoxidation of \( {\hbox{Q}}_{\hbox{A}}^ - \) by QB as in thylakoid membranes (Pospišil and Tyystjärvi 1999; Putrenko et al. 1999; Wijn and van Gorkom 2001). The middle phase is ascribed for the reoxidation of \( {\hbox{Q}}_{\hbox{A}}^ - \) in PSII centers with an empty QB pocket where the diffusion time of PQ to its binding site should be considered. The slow phase is associated with the recombination of \( {\hbox{Q}}_{\hbox{A}}^ - \) with the S2 and/or S3 states of the Mn-cluster or with cytochrome b559 in inactive PSII centers (Putrenko et al. 1999; Wijn and van Gorkom 2001). Another interpretation of the slow phase in thylakoid membranes is the reoxidation of \( {\hbox{Q}}_{\hbox{A}}^ - \) in PSII centers with an empty QB pocket following the binding of PQ migrating from a slow pool (Pospišil and Tyystjärvi 1999). The amplitude and half-time of each component are shown in Table 1. The half-times greatly increased and a 25% reduction of the relative amplitude of the fast phase was also observed with Ni2+ showing that \( {\hbox{Q}}_{\hbox{A}}^ - \) reoxidation was much slower in the presence of the metal cation. It is observed in Fig. 8A that the amplitude of the total fluorescence rise induced by the flash declined with Ni2+. In Fig. 8B, typical traces of fluorescence decays show that the loss in amplitude following Ni2+ treatment (trace 2) was greatly recovered with addition of CaCl2 (trace 3).

(A) Chl fluorescence decay kinetics of PSII submembrane fractions alone (trace 1) or treated with 0.1 (trace 2) or 0.2 (trace 3) mmol NiCl2/mg Chl. The solid lines represent the curve fit obtained using a three-exponential decay function as mentioned in “Materials and methods”. (B) Redox kinetics of QA measured as Chl fluorescence in control samples (trace 1) or in samples incubated with 0.1 mmol NiCl2/mg Chl alone (trace 2) or together with 0.2 mmol CaCl2 (trace 3). Upward arrows indicate the application of the single turn-over saturating white flash. Each trace is the average of nine independent measurements
Discussion
Previous reports indicated that relatively high concentrations of Ni2+ (5–10 mM) produced a significant inhibitory effect in chloroplasts (Tripathy et al. 1983; Mohanty et al. 1989; Szalontai et al. 1999) or in green alga (El-Sheekh 1993). In the green alga Haematococcus lacustris, 0.1 mM Ni2+ already affected F v/F m measured at a Chl concentration of 15 μg Chl ml−1 but only after several days of exposure to the toxic cations (Xyländer and Braune 1994). In the present study, we used PSII submembrane fractions where both stromal and lumenal sides of the photosystem are more readily accessible to inhibitors compared with whole organisms or isolated thylakoid membranes. A 10-min incubation of PSII submembrane fractions at a concentration of 10 μg Chl ml−1 in the presence of 3.5 mM Ni2+ was able to decrease oxygen evolution activity by about 50% (Fig. 1). Inhibition of electron transport is also shown by a decline of the fluorescence induction components in correspondence with a reduced F v/F m with Ni2+. The O-J phase is associated with the redox state of the primary quinone acceptor QA (Pospišil and Dau 2000). The I-P rise in the fluorescence induction traces of PSII submembrane fractions was shown to coincide with the reduction of the PQ pool (Pospišil and Dau 2000) thus lowering thermal quenching of variable Chl fluorescence by oxidised PQ (Samson et al. 1999). The decline in F v/F m together with the amplitudes of the O-J and I-P phases of the FI curve indicates that the OEC failed to provide electrons for PSII to reduce adequately the quinone acceptors of the photosystem in the presence of Ni2+ thus decreasing the maximal fluorescence yield. Further, the half-time of the I-P phase increased proportionally with the inhibition of the oxygen evolution activity (Pospišil and Dau 2000). Therefore, its dependency on Ni2+ concentration is consistent with the inhibition of oxygen evolution shown in Fig. 1.
The inhibitory action of Ni2+ was closely related to the removal of the extrinsic polypeptides of 17 and 24 kDa that are associated with the OEC of PSII (Seidler 1996). In fact, depletion of these polypeptides occurs at concentrations similar to inhibition of oxygen evolution. Several toxic divalent cations were shown by Fourier transform infrared spectroscopy to form metal–protein complexes due to their association with C=O, C–N, and/or SH groups of amino acids and to induce protein conformational transitions in PSII submembrane fractions (Nahar and Tajmir-Riahi 1996). Such interaction with the extrinsic polypeptides could have produced conformational changes that provoked their dissociation from the membrane and resulted in strong inhibition of electron transport activity.
The removal of the two polypeptides of 17 and 24 kDa locates the inhibitory site directly at the level of the OEC. This interpretation is supported by the antagonistic effect between Ca2+ and Ni2+ (Fig. 2) and the loss of two Mn atoms from the OEC. Calcium is a well-known essential cofactor for oxygen evolution (Seidler 1996). From 18O isotope exchange studies, it was recognized that a calcium ion is bound to one of the substrate-water molecule of the OEC (Hendry and Wydrzynski 2003) being located at only about 3.5 Å from the Mn cluster (Cinco et al. 1998). Recent structural data obtained from X-ray spectroscopy of the cyanobacterium Thermosynechoccocus elongatus photosystem II at 3.5 and 3.0 Å resolution support the presence of a Ca2+ ion in direct interaction with the Mn cluster (Iwata and Barber 2004; Loll et al. 2005). The two extrinsic polypeptides of 17 and 24 kDa modulate the affinity of the Ca2+ binding site (Ghanotakis et al. 1984; Adelroth et al. 1995). Hence, Ca2+ is lost when these two polypeptides forming a barrier between the lumen and the calcium-binding site are released from the OEC (Yocum 1991). Ca2+ is needed for the proper advancement of the S states of the OEC (Boussac and Rutherford 1988). It is clear that inhibition of PSII electron transport by Ni2+ is associated with the removal of Ca2+ from its site in the OEC following removal of the two extrinsic polypeptides and competition of Ni2+ at the Ca2+ binding site.
Indeed, the most competitive interaction found between Ni2+ and Ca2+ indicates that the two cations interact at the same site in the OEC. Our results are consistent with a previous report using PSII submembrane fractions depleted in Ca2+, and 17 and 24 kDa polypeptides, where a competitive inhibition of oxygen evolution with a small component of mixed inhibition was observed for cations (including Ni2+) with relatively small ionic radii relative to Ca2+ (Vrettos et al. 2001). Exogenously added CaCl2 can restore the oxygen-evolving activity in preparations depleted in 17 and 24 kDa polypeptides (Nakatani 1984). Thus in the presence of excess CaCl2, the PSII activity is largely protected even when the polypeptides are released by Ni2+.
It was previously proposed that Ni2+ inhibits PSII electron transport activity at the level of QB (Mohanty et al. 1989; El-Sheekh 1993) but at the same time, the quenching of variable Chl fluorescence following Ni2+ treatment indicated an inhibition of oxygen evolution (Tripathy et al. 1981; Singh et al. 1989; Mohanty et al. 1989; El-Sheekh 1993). The data presented here using PSII preparations clearly demonstrate an inhibitory site for Ni2+ at the OEC. However, a secondary action at other sites cannot be ruled out. An active site on the acceptor side of PSII was deduced for Cu2+ and Cd2+ from studies involving fluorescence and EPR spectroscopy (Jegerschöld et al. 1995; Sigfridsson et al. 2004). The flash-induced Chl fluorescence decay measurements presented here indicate that reoxidation of \( {\hbox{Q}}_{\hbox{A}}^ - \) is greatly retarded with Ni2+. The above could indicate an inhibitory site between QA and QB. However, Putrenko et al. (1999) have shown that the half-time of \( {\hbox{Q}}_{\hbox{A}}^ - \) reoxidation was strongly increased in NaCl-treated PSII submembrane fractions that are depleted of the two extrinsic polypeptides of 17 and 24 kDa and Ca2+. This was explained by a transmembrane conformational change that modifies the acceptor side of PSII reflecting a long-range allosteric coupling with the OEC. Such alteration of the acceptor side is also expected in Ni2+-treated PSII as we have shown that the action of this metal is very much similar to NaCl treatment in terms of the removal of extrinsic polypeptides. This interpretation is supported by the fact that CaCl2 restored the fluorescence kinetics in Ni2+-treated PSII preparations (Fig. 8B).
Ni2+ treatment also caused a decline of the amplitude of the fluorescence rise induced by a single turn-over flash (see Fig. 8B). This apparent loss of fluorescence indicates that there is a fast decline of fluorescence that occurs before the start of the decay measurement at 100 μs. This decline is likely due to recombination between \( {\hbox{Q}}_{\hbox{A}}^ - \) and P680+. PSII centers with P680+ are known to have a fluorescence yield close to F 0 (Deprez et al. 1983). However, the accumulation of P680+ after a single flash illumination is only observed when electron transfer between the primary electron donor TyrZ and P680 is inhibited (Britt 1996). It was shown that the loss of Ca2+ ion alone is insufficient to decrease the rate of reduction of P680+ by TyrZ (Stevens and Lukins 2003). However, decreased rates of electron transfer from TyrZ to P680 in PSII centers that have lost the two extrinsic polypeptides of 17 and 24 kDa together with Ca2+ is not surprising because structural data suggested that the Ca2+ site is located in close proximity with TyrZ (Iwata and Barber 2004; Loll et al. 2005). Further, addition of CaCl2 restored the amplitude of the fluorescence rise (Fig. 8B). Thus, the presence of Ni2+ may influence the specific orientation of TyrZ or hinder its deprotonation that is needed for electron transfer to P680+ (Christen and Renger 1999).
Several other toxic divalent cations such as Cu2+, Cd2+, Hg2+, Zn2+, and Pb2+, were shown to release the extrinsic polypeptides of PSII to various extents (Rashid et al. 1994; Bernier and Carpentier 1995; Jegerschöld et al. 1995; Yruela et al. 2000). However, the negative effect of Ni2+ in PSII is most closely related to the action of Zn2+. The latter also affected PSII with a 50% inhibition at a concentration of about 1 mM, released the same two extrinsic polypeptides together with two Mn atoms, and its inhibition was relieved in the presence of Ca2+ (Miller 1985; Rashid et al. 1991, 1994). Exposure of plants to nickel greatly affects photosynthesis (Tripathy et al. 1981; Mohanty et al. 1989; Krupa et al. 1993). However the exact nickel concentration in leave tissues is difficult to determine. It was reported that nickel content can increase by several folds upon plant exposure to nickel chloride (Gopal et al. 2002). Also, nickel was found to accumulate in the chloroplast and concentrate in the form of electron-dense precipitates on the chloroplast membranes (Veeranjaneyulu and Das 1982; Molas 1997). Therefore, there is a possibility that the effects found in this study using isolated PSII membranes also occur in vivo.
Abbreviations
- Chl:
-
chlorophyll
- FI:
-
fluorescence induction
- F 0 :
-
initial fluorescence
- F v :
-
variable fluorescence
- F m :
-
maximal fluorescence
- MES:
-
2-(N-morpholino)ethanesulfonic acid
- OEC:
-
oxygen-evolving complex
- P680:
-
primary electron donor of photosystem II
- PQ:
-
plastoquinone
- PSII:
-
photosystem II
- QA :
-
primary quinone acceptor
- QB :
-
secondary quinone acceptor
References
Adelroth P, Lindberg K, Andreasson LE (1995) Studies of Ca2+ binding in spinach photosystem II using 45Ca2+. Biochemistry 34:9021–9027
Babani F, Lichtenthaler HK (1996) Light-induced and age-dependent development of chloroplasts in etiolated barley leaves as visualized by determination of photosynthetic pigments, CO2 assimilation rates and different kinds of chlorophyll fluorescence ratios. J Plant Physiol 148:555–566
Berthold DA, Babcock GT, Yocum CF (1981) A highly resolved oxygen-evolving photosystem II preparation from spinach thylakoid membranes. EPR and electron transport properties. FEBS Lett 134:231–234
Bernier M, Carpentier R (1995) The action of mercury on the binding of the extrinsic polypeptides associated with the water oxidizing complex of photosystem II. FEBS Lett 360:251–254
Boisvert S, Joly S, Carpentier R (2006) Quantitative analysis of the experimental O-J-I-P chlorophyll fluorescence induction kinetics. Apparent activation energy and origin of each kinetic step. FEBS J 273:4770–4777
Boussac A, Rutherford AW (1988) S-state formation after Ca2+ depletion in the photosystem II oxygen-evolving complex. Chem Scr 28A:123–126
Britt RD (1996) Oxygen evolution. In: Ort DR, Yocum F (eds) Oxygenic photosynthesis: the light reactions. Kluwer Academic Publishers, Dordrecht, The Netherlands, pp 137–164
Carpentier R (2002) The negative action of toxic divalent cations on the photosynthetic apparatus. In: Pessarakli M (ed) Handbook of plant and crop physiology. Marcel Dekker, New York, pp 764–772
Christen G, Renger G (1999) The role of hydrogen bonds for the multiphasic P680+ reduction by YZ in photosystem II with intact oxygen evolution capacity. Analysis of kinetic H/D isotope exchange effects. Biochemistry 38:2068–2077
Cinco RM, Robblee JH, Rompel A, Fernadez C, Yachandra VK, Sauer K, Klein MP (1998) Strontium EXAFS reveals the proximity of calcium to the manganese cluster of oxygen-evolving photosystem II. J Phys Chem B 102:8248–8256
Clijsters H, van Asche P (1985) Inhibition of photosynthesis by heavy metals. Photosynth Res 7:731–740
Deprez JA, Dobek NE, Geacintov G, Paollotin G, Breton J (1983) Probing fluorescence induction in chloroplasts on a nanosecond time scale utilizing picosecond laser pulse pairs. Biochim Biophys Acta 725:444–454
De Wijn R, van Gorkom J (2001) Kinetics of electron transfer from QA to QB in photosystem II. Biochemistry 40:11912–11922
El-Sheekh MM (1993) Inhibition of photosystem II in the green alga Scenedesmus obliguus by nickel. Biochem Physiol Pflanzen 188:363–372
Ghanotakis DF, Topper JN, Babcock GT, Yocum CF (1984) Water-soluble 17-kDa and 23-kDa polypeptides restore oxygen evolution activity by creating a high-affinity binding site for Ca2+ on the oxidizing side of photosystem II. FEBS Lett 170:169–173
Gopal R, Mishra KB, Zeeshan M, Prasad SM, Joshi MM (2002) Laser-induced chlorophyll fluorescence spectra of mung plants growing under nickel stress. Curr Sci 83:880–884.
Iwata S, Barber J (2004) Structure of photosystem II and molecular architecture of the oxygen-evolving center. Curr Opin Struct Biol 14:447–453
Hendry G, Wydrzynski T (2003) 18O isotope exchange measurements reveal that calcium is involved in the binding of one substrate-water molecule of the oxygen-evolving complex in photosystem II. Biochemistry 42:6209–6217
Jegerschöld C, Arellano JB, Schröder WP, van Kan PJM, Barón M, Styring S (1995) Copper(II) inhibition of electron transfer through photosystem II studied by EPR spectroscopy. Biochemistry 34:12747–12754
Krupa Z, Siedleka A, Maksymiec W, Baszynski T (1993) In vivo response of photosynthetic apparatus of Phaseolus vulgaris L. to nickel toxicity. J Plant Physiol 142:664–668
Lazár D (1999) Chlorophyll fluorescence induction. Biochim Biophys Acta 1412:1–28
Lazár D (2006) The polyphasic chlorophyll a fluorescence rise measured under high intensity of exciting light. Funct Plant Biol 33:9–30
Loll B, Kern J, Saenger W, Zouni A, Biesiaka J (2005) Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature 438:1040–1044
Miller M (1985) The release of polypeptides and manganese from oxygen-evolving photosystem II preparations following zinc-treatment. FEBS Lett 189:355–360
Mohanty N, Vass J, Demeter S (1989) Impairment of photosystem II activity at the level of secondary quinone electron acceptor in chloroplasts treated with cobalt, nickel and zinc ions. Physiol Plant 76:386–390
Molas J (1997) Changes in morphological and anatomical structure of cabbage (Brassica oleacera L.) outer leaves and in ultrastructure of their chloroplasts caused by an in vitro excess of nickel. Photosynthetica 34:513–522
Nahar S, Tajmir-Riahi HA (1996) Complexation of heavy metal cations Hg, Cd and Pb with proteins of PSII: evidence for metal–sulfur binding and protein conformational transition by FTIR spectroscopy. J Coll Interface Sci 178:648–656
Nakatani HY (1984) Photosynthetic oxygen evolution does not require the participation of polypeptides of 16 and 24 kilodaltons. Biochem Biophys Res Commun 120:299–304
Ono T, Noguchi T, Nakajima Y (1995) Characteristic changes of function and structure of Photosystem II during strong-light photoinhibition under aerobic conditions. Biochim Biophys Acta 1229:239–248
Pospišil P, Dau H (2000) Chlorophyll fluorescence transients of photosystem II membrane particles as a tool for studying photosynthetic oxygen evolution. Photosynth Res 65:41–52
Pospišil P, Dau H (2002) Valinomycin sensitivity proves that light-induced thylakoid voltages result in millisecond phase of chlorophyll fluorescence transients. Biochim Biophys Acta 1554:94–100
Pospišil P, Tyystjärvi E (1999) Molecular mechanism of high-temperature-induced inhibition of acceptor side of photosystem II. Photosynth Res 62:55–66
Putrenko II, Vasil’ev S, Bruce D (1999) Modulation of flash-induced photosystem II fluorescence by events occurring at the water oxidizing complex. Biochemistry 38:10632–10641
Rashid A, Bernier M, Pazdernick L, Carpentier R (1991) Interaction of Zn2+ with the donor side of photosystem II. Photosynth Res 30:123–130
Rashid A, Camm EL, Ekramoddoulah AKM (1994) Molecular mechanism of action of Pb2+ and Zn2+ on water oxidizing complex of photosystem II. FEBS Lett 350:296–298
Samson G, Prasil O, Yaakoubd B (1999) Photochemical and thermal phases of chlorophyll a fluorescence. Photosynthetica 37:163–182
Seidler A (1996) The extrinsic polypeptides of photosystem II. Biochim Biophys Acta 1277:35–60
Sheoran IS, Singal HR, Singh R (1990) Effects of cadmium and nickel on photosynthesis and the enzymes of the photosynthetic carbon reduction cycle in pigeon pea (Cajanus cajan L.). Photosynth Res 23:345–351
Singh DP, Khare P, Bisen PS (1989) Effect of Ni2+, Hg2+ and Cu2+ on growth, oxygen evolution and photosynthetic electron transport in Cylindrospermum IU 942. J Plant Physiol 134:406–412
Stevens GB, Lukins PB (2003) Effect of Ca2+ and EGTA on P680+ reduction kinetics and O2 evolution of photosystem II. Biochim Biophys Acta 1605:21–34
Sigfridsson KGV, Bernat G, Mamedov F, Styring S (2004) Molecular interference of Cd2+ with photosystem II. Biochim Biophys Acta 1659:19–31
Singh DP, Khare P, Singh PS (1989) Effect of Ni2+, Hg2+ and Cu2+ on growth, oxygen evolution and photosynthetic electron transport in Cylindrospermum IU 942. J Plant Physiol 134:406–412
Szalontai B, Horvath LI, Debreczeny M, Droppa M, Horvath G (1999) Molecular rearrangements of thylakoids after heavy metal poisoning, as seen by Fourier transform infrared (FTIR) and electron spin resonance (ESR) spectroscopy. Photosynth Res 61:241–252
Tripaty BC, Bhatia B, Mohanty P (1981) Inactivation of chloroplast photosynthetic electron transport activity by Ni2+. Biochim Biophys Acta 638:217–224
Tripaty BC, Bhatia B, Mohanty P (1983) Cobalt ions inhibit electron transport activity of photosystem II without affecting photosystem I. Biochim Biophys Acta 722:88–93
Veeranjaneyulu K, Das VSR (1982) Intrachloroplast localization of 65Zn and 63Ni in Zn-tolerant plant (Ocimum basilicum Benth.). J Exp Bot 137:1161–1165
Vrettos JS, Stone DA, Brudvig GW (2001) Quantifying the ion selectivity of the Ca2+ site in photosystem II: evidence for direct involvement of Ca2+ in O2 formation. Biochemistry 40:7937–7945
Xyländer M, Braune W (1994) Influence of nickel on the green alga Haematococcus lacustris Rostafinski in phases of its life cycle. J Plant Physiol 144:86–93
Yocum CF (1991) Calcium activation of photosynthetic water oxidation. Biochim Biophys Acta 1059:1–15
Yruela I, Alfonso M, Barón M, Picorel R (2000) Copper effect on the protein composition of photosystem II. Physiol Plant 110:551–557
Zhu X-G, Govindjee, Baker NR, deSturler E, Ort DR, Long SP (2005) Chlorophyll a fluorescence induction kinetics in leaves predicted from a model describing each discrete step of excitation energy and electron transfer associated with photosystem II. Planta 223:114–133
Acknowledgements
This work was funded by the Natural Science and Engineering Research Council of Canada. DJ was supported by a postgraduate fellowship from Fonds Québécois de la Recherche sur la Nature et les Technologies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Boisvert, S., Joly, D., Leclerc, S. et al. Inhibition of the oxygen-evolving complex of photosystem II and depletion of extrinsic polypeptides by nickel. Biometals 20, 879–889 (2007). https://doi.org/10.1007/s10534-007-9081-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-007-9081-z