
Abstract
Recently, natural tyrosinase inhibitors have gained attention in clinical cosmetology research. In this study, the enzymatic hydrolysis of Pinctada martensii meat by protease from Bacillus licheniformis, 401 peptides with tyrosinase inhibitory were identified after isolated by ultrafiltration and Sephadex G-15 from the fraction F4. The peptide effects on the tyrosinase activity and structure were evaluated using molecular docking. Three synthetic peptides classified as W1 (WDRPKDDGGSPIK), W2 (DRGYPPVMF), and W3 (SGGGGGGGLGSGGSIRSSY), which had the lowest binding energies were selected for in vitro synthesis and biological activity investigation. The W3 peptide (5 mg/mL) had the highest tyrosinase activity, SPF, DPPH, and ABTS clearance values, and total antioxidant capacity. W3 did not affect the survival rate of mouse melanoma B16-F10 cells (1.0–5.0 mg/mL) but decreased the melanin content. Hence, W3 could be suitable for multifunctional tyrosinase inhibition and provides a novel method to use marine organisms as natural tyrosinase inhibitor sources.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As one of the most widely distributed pigments, melanin protects the human skin from damage by absorbing ultraviolet (UV) light and removing reactive oxygen species (Kim and Uyama 2005). However, the overproduction of melanin, caused by chloasma generation, senile plaques, and photochemical damage (Gou et al. 2017), threatens skin health. Tyrosinase (EC 1.14.18.1) is a crucial enzyme involved in melanin synthesis, which oxidizes l-tyrosine to l-3,4-dihydroxyphenylalanine (l-DOPA) through monophenolase and bisphenolase, and is further oxidized into dopoquinone during polymerization (Feng et al. 2021). These processes result in melanin formation (Yu and Fan 2021). Thus, tyrosinase is a critical element that can potentially be used to inhibit melanin synthesis.
Currently, arbutin and kojic acid are representative antimelanogenic agents used in the cosmetic industry for antityrosinase activity (Han et al. 2011). However, their safety issues in pharmaceutical products have been previously reported (Burnett et al. 2010; Jeon et al. 2019; Kim et al. 2019). Arbutin has remained in use in the cosmetic industry; however, the European Union has not used other hydroquinones in skin-lightening formulations since 2001 (O’Donoghue 2006). This suggests that other natural products with antimelanogenic activity have already been identified and used to protect human skin from melanogenesis (Heriniaina et al. 2018). Owing to this, plant extracts of stem bark, methanol, and ethyl acetate from Alnus cordata, Momordica charantia, and Calendula officinalis flowers, respectively, have gained increasing attention, especially in efficacy comparison studies (Park et al. 2019), to find depigmenting agents with antimelanogenic properties and replace arbutin and kojic acid (Lee et al. 2019). Therefore, the development of natural tyrosinase inhibitors, such as bioactive peptides, has attracted considerable attention in clinics and cosmetics (Park et al. 2014; Pilevneli and Konuklugil 2020). For example, Ochiai et al. (2016) developed a technique for producing tyrosinase inhibitory peptides from rice bran, a by-product of rice. They treated the rice bran protein simultaneously with chymotrypsin and trypsin, leading to the generation of a substantial quantity of peptides. Subsequently, they employed a multi-step purification scheme to isolate six tyrosinase inhibitory peptides from the hydrolysate, and their amino acid sequences were determined. Out of these, three peptides demonstrated remarkable inhibitory activity against the tyrosinase-mediated monophenolase reaction. Ge et al. (2023) aimed to isolate and analyze tyrosinase inhibitory peptides from the enzymatic digestion products of royal jelly. They optimized the enzymatic hydrolysis of royal jelly using single-factor experiments and response surface methodology. The component with the highest tyrosinase inhibitory activity was purified and isolated through gel filtration chromatography. Subsequently, three highly active tyrosinase inhibitory peptides were screened using LC–MS/MS and molecular docking analysis.
The extreme oceanic environment, characterized by factors such as pressure, light, temperature, and salinity, plays a significant role in the production of a diverse range of functional bioactive compounds by marine organisms (Yang et al. 2022). Among these compounds, proteinaceous substances, including proteins, peptides, and amino acids, hold particular importance. Additionally, certain fish and shellfish species, known for their high protein content, serve as excellent sources for generating bioactive peptides derived from proteins (Hao et al. 2022; Harnedy and FitzGerald 2012). These peptides effectively improve osteoblast differentiation in human bone marrow-derived mesenchymal stem cells (hBMMSCs) by upregulating the canonical Wnt/β-catenin signaling, preventing ovariectomy-induced bone loss (Oh et al. 2020). Pinctada martensii is the primary marine-cultured shellfish in the southern coastal areas of Guangxi and Guangdong, China (Zou et al. 2015). Its protein content is significantly higher than other shellfish species such as oysters and mussels (Xiaowei et al. 2004). Hence, more material can be used to continuously produce bioactive peptides with antihypertensive, antitumor, antibacterial, and immune-enhancing properties (Hao 2007; Wang et al. 2014). However, this results in a critical shortage of shellfish supply (Liu et al. 2020). Efficiently utilizing this resource is critical since a significant fraction of the obtained meat can only be used as a feed or dry product and is otherwise discarded.
In this study, the tyrosinase inhibition peptides were obtained from P. martensii meat due to alkaline protease hydrolysis of Bacillus licheniformis and purification using ultrafiltration and Sephadex G-15 column. Several fractions were collected and lyophilized to be further identified using the reversed-phase liquid chromatography–mass spectrometry (RPLC–MS). Molecular docking simulation was then applied to the selected probable tyrosinase inhibitory peptides. Subsequently, we validated the activity of the synthesized peptides through in vitro caseinase inhibition assay, antioxidant activity measurement, and determination of sun protection factor (SPF). In this study, we screened peptides with caseinase inhibitory activity from the P. martensii meat coruscus, providing a new direction for the utilization of discarded P. martensii meat.
Materials and methods
Materials and chemicals
P. martensii meat was acquired from Guangxi Beihai Seiko Marine Co., Ltd (Guangxi, China). The P. martensii meat was transported to the laboratory under frozen conditions. After the meat was washed three times with deionized water and drained, it was aliquoted and stored in a −20 °C freezer. All kinds of protease were purchased from Sigma-Aldrich (St. Louis, MO, USA). l-Tyrosine, Sephadex G-15 medium, trypsin-ethylenediaminetetraacetic acid (EDTA) digestive solution, phosphate-buffered saline (PBS), penicillin, streptomycin, and tyrosinase activity test kit were purchased from Beijing Solarbio Technology Co., Ltd (Beijing, China). l-DOPA and mushroom tyrosinase (1240 U/mg) were obtained from Shanghai Yuanye Biotechnology Co., Ltd (Shanghai, China). Murine melanoma B16-F10 cells were obtained from the Stem cell bank of the Chinese Academy of Sciences (Shanghai, China). Gibco Australian fetal bovine serum (FBS) and Dulbecco’s modified Eagle medium (DMEM) high glucose medium were purchased from Thermo Fisher Technology (China) Co., Ltd (Shanghai, China). Cell Counting Kit-8 (CCK-8) was obtained from Med Chem Express (Monmouth Junction, NJ, USA). The Mouse melanin ELISA kit was bought from Shanghai ML Bio Biotechnology Co., Ltd (Shanghai, China). Analytical grade chemicals and reagents were used in this study.
Screening of the targeted protease
Bacillus licheniformis protease, neutral protease, alpha-chymotrypsin, papain, trypsin, flavourzyme, and pepsin or compound proteinase were applied to enzymatic hydrolysates of P. martensii meat (PMHs). The fresh tissues of P. martensii were boiled for 2 min at 100 °C and homogenized with a 1:4 (w/w) ratio of deionized water. A 5000 U/g enzyme was then added to hydrolyze the protein dispersion and the different proteases in the following conditions: pH 7.5 at 60 °C, pH 7.0 at 50 °C, pH 7.0 at 37 °C, pH 7.0 at 50 °C, pH 8.0 at 50 °C, pH 6.0 at 50 °C, pH 3.0 at 37 °C, pH 8.0 at 50 °C, respectively, for 4 h. Thereafter, the resultant hydrolysates were immediately boiled for 10 min and centrifuged at 8600×g for 15 min (4 °C). The supernatant was collected, adjusted to pH 7.0, and stored at −20 °C for further analysis. To obtain more representative results than a single factor test, the reaction conditions, such as the time, temperature, enzyme addition, pH, and material to water ratio, were selected as the main factors affecting enzymatic hydrolysis (temperature options: 55 °C, 60 °C, 65 °C; time options: 3 h, 4 h, 5 h; enzyme dosage options: 1000 U/g, 3000 U/g, 5000 U/g; pH options: 7, 7.5, 8.0; substrate-to-water ratio options: 1:4, 1:6, 1:8). SPSS 22.0 software was used to conduct one-way analysis of variance (ANOVA) and Duncan’s multiple comparisons (p < 0.05).
Determination of tyrosinase inhibitory activity
The method to determine tyrosinase inhibitory activity was based on previous studies (Yap and Gan 2021), but with slight modifications. Samples of 40 µL and 140 µL of 50 mM phosphate buffer (PBS, pH 6.8) containing 40 µL tyrosinase (500 U/mL) were added to a 96-well plate and incubated at 37 °C for 10 min. A 20 µL l-tyrosine (8.4 mM) was subsequently added and incubated for 5 min at 37 °C. The plate was then measured at 475 nm wavelength in a microplate reader (TECAN, Infinite M200 PRO, Switzerland). The samples without l-tyrosine and with arbutin were used as the blank and positive controls, respectively. The tyrosinase inhibitory activity was calculated as follows:
Determination of peptide yield
The peptide yield of hydrolysates was determined by measuring the peptide content in 10% trichloroacetic acid (TCA) (Yap and Gan 2020), in which enzymatic hydrolysates and TCA (10%, w/v) in equal volumes were thoroughly mixed and left to stand for 10 min at 30 °C. The mixture was then centrifuged at 6000×g for 15 min. The supernatant was mixed with biuret in a 1:4 volume ratio, and the reactants were measured at 540 nm after being left to stand for 60 min at 30 °C, with water as the blank control. The peptide concentration was calculated using a standard curve prepared with a bovine serum albumin standard. Peptide yield (%) was calculated using the equation:
Purification of peptides from PMHs
The hydrolysates of P. martensii meat (PMHs) were fractioned using ultrafiltration membranes (Millipore, USA) with a molecular cutoff of 3 kDa and 5 kDa to obtain three fractions: PMHs-1 (< 3 kDa), PMHs-2 (3–5 kDa) and PMHs-3 (> 5 kDa). Each fraction was freeze-dried and tested for tyrosinase inhibitory activity at 1.0 mg/mL concentration.
Meanwhile, the lyophilized hydrolysate with the highest tyrosinase inhibitory activity was dissolved in ultrapure water and further purified using a 0.22-μm filter. The filtrate (1.0 mL, 10 mg/mL) was then loaded onto a Sephadex G-15 column (1.6 × 80 cm, Shanghai Xiamei Biochemical Technology Development Co., Ltd, Shanghai, China) connected to the AKTA protein-purification system. The column was eluted and equilibrated with ultrapure water at a flow rate of 1.0 mL/min. All eluate fractions were monitored at 280 nm, and the measurements with significant inhibitory activity were collected for further analysis.
Identification, molecular docking, and synthesis of tyrosinase inhibitory peptides
The amino acid sequence and fraction with the highest tyrosinase inhibitory activity were analyzed using an RPLC–MS instrument (Thermo Fisher Scientific, USA). The peptide samples were dissolved in 0.1% TFA and desalinized using a 10 μL matrix solution (60% acetonitrile (ACN) solution containing 0.1% TFA). The freeze-dried peptides were mixed with 0.1% formic acid and 5% ACN. The supernatant was analyzed using the RPLC–MS following its centrifugation at 13,500 rpm for 20 min at 4 °C. Buffer A (0.1% formic acid) and buffer B (0.1% formic acid and 80% ACN) were used as the mobile phases in the gradient elution with the following parameters: (1) MS: scan range (m/z) = 350–1550; resolution = 120,000; AGC target = 4e5; (2) MS/MS: resolution = 30,000; AGC target = 1e5; maximum injection time = 100 ms; and top N = 20; NCE/steeped NCE = 32. Peptide sequencing was conducted by processing the MS/MS data using the PEAKS software and manual calculation. Data was matched with NCBI (https://www.ncbi.nlm.nih.gov/), with an access date of May 18, 2021.
Additionally, the molecular docking was performed for sequences with a confidence interval of > 45.0 to obtain the X-ray crystal structure of mushroom tyrosinase (PDB ID:2Y9X) from the RCSB Protein Data Bank. The three-dimensional structures of the selected peptides were generated by PepFold3 web server (Lamiable et al. 2016), and all parameters used were the default settings of the web page. The Autodock 4.2 software (Langeswaran et al. 2019) was used to remove water molecules and add polar hydrogen molecules of mushroom tyrosinase and peptides. In contrast, the HPEPDOCK server (Yan et al. 2020) was used to bind peptide-protein by fast modeling peptide conformations and global sampling of binding orientations. Lastly, the HPEPDOCK submission form and the tyrosinase structure file were uploaded as receptor inputs, whereas the peptide structure file was uploaded as the peptide input. The protein–peptide docking solutions were ranked using a docking score, wherein the cluster with the lowest score was considered the most likely conformation. Finally, the PRODIGY web server (Vangone and Bonvin 2017) and LigPlot+ software (Yap and Gan 2021) were used to predict and visualize the binding affinity of the protein–peptide complex and binding interactions, respectively. To evaluate the tyrosinase inhibitory activity of screened peptides, three monophenolase inhibitor peptides (WDRPKDDGGSPIK (MW = 1470.58 Da), DRGYPPVMF (MW = 1081.25 Da), and SGGGGGGGLGSGGSIRSSY (MW = 1569.59 Da), were subjected to chemical synthesis by the Sangon Biotech (Shanghai, China), resulting in three classifications, namely: W1, W2, and W3, respectively.
Determination of the multifunctional activity of synthetic peptides in vitro
Determination of sun protection factor (SPF)
The SPF of synthetic peptides was determined as described in a previous study (Yap and Gan 2021). The multifunctional microplate reader was set to scan the samples (100.0 µL) from 220–400 nm with 5 nm increments. The SPF values of the peptide samples (5.0 mg/mL) were then calculated using the Mansur equation:
where CF is the correction factor of 10, EE is the erythemal effect spectrum, I is the solar intensity spectrum, and Abs is the absorbance of the sample. The values of EE × I were constants, specifically the wavelengths of 290, 295, 300, 305, 310, 315 and 320 nm were 0.0150, 0.0817, 0.2874, 0.3278, 0.1864, 0.0839 and 0.0180, respectively (Dutra et al. 2004).
Determination of antioxidant activity
Determination of 2,2-diphenyl-1-picrylhydrazyl (DPPH) free radical scavenging
The DPPH free radical scavenging potential of peptides was measured as described in a previous study (Deng et al. 2020), but with some modifications. First, 100 µL of 0.1 mM DPP prepared in ethanol was added to a 50 µL peptide sample (5 mg/mL) and incubated at 37 °C for 30 min in a dark setting. The absorbance of the mixture was monitored at 517 nm using a multifunctional microplate reader (TECAN, Infinite M200 PRO), using glutathione (GSH) as the positive control. The DPPH free radical scavenging activity was calculated as follows:
where A0 is the absorbance of the sample and DPPH, A1 is the absorbance of sample and ethanol without DPPH, and A2 is the absorbance of the control (water) without sample.
Determination of 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic) acid (ABTS) free radical scavenging
The reference method was slightly modified (Yap and Gan 2021), wherein the 7 mM 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic) acid (ABTS) and 2.6 mM ammonium persulfate solutions were subjected to reaction at room temperature without light for 12 h. Thereafter, the mixed reagent was diluted with 80% ethanol to an absorbance of 0.700 ± 0.005 at 734 nm. A 195 µL of diluted reagent was then added to the 5 µL peptide samples solution. The absorbance value of the sample was measured at 734 nm immediately after the 6 min light-shielding reaction. Using glutathione (GSH) as the positive control, the free radical scavenging activity of ABTS was calculated as follows:
where A0 is the blank control, i.e., 195 µL diluted ABTS reagent + 5 µL double distilled water, and A1 is the 195 µL diluted ABTS reagent + 5 µL sample solution.
Determination of total antioxidant capacity (FRAP)
As antioxidants reduce Fe3+-tripyridine triazine (Fe3+-TPTZ) and produce blue Fe2+-TPTZ in an acidic environment, the total antioxidant capacity of the sample was measured using the kit (FRAP) to determine the blue Fe2+-TPTZ at 590 nm and obtained the total antioxidant capacity in the sample. The color-developing solution was prepared according to the instructions, and the reaction solutions were added to the 96-well plate, as shown in Table 1 below. After mixing the reactions for 10 min at 25 °C, the absorbance value was measured at 590 nm using Trolox to design the standard curve. Using GSH as the positive control, the total antioxidant capacity was calculated as follows:
Murine melanoma B16-F10 cell model
B16-F10 cells with logarithmic growth were digested by trypsin–EDTA solution, wherein the cell density was adjusted to 1 × 105 pieces/mL (Kim et al. 2017). The cell suspension (100 μL) was inoculated in a 96-well plate μL/well, and the orifice plate was placed inside the carbon dioxide incubator to undergo cell adhesion. After the 24 h inoculation, the culture medium was carefully aspirated, and the different concentrations of sample solutions (100 μL/well) were added and cultured for another 24 h. The Cell Counting Kit-8 (CCK-8) method using the mouse melanin ELISA was used to determine the cell survival rate, whereas the tyrosinase activity test kit was used for the melanin content and tyrosinase activity.
Kinetic analysis of the inhibition of tyrosinase
The activity of the monophenolase reaction was determined by measuring the formation rate of dophenolamine. In the presence of peptide samples, the kinetic study (apparent Km and Vmax) of tyrosinase toward l-tyrosine was determined as described previously (Dutra et al. 2004). The initial velocity V of the reaction was determined using Bill Lambert’s Law:
where \(\Delta {\text{A}}/t\) is the rate of change of absorbance or the slope of the reaction, ε is the molar extinction coefficient of dopachrome (3700 M−1/cm), and l is the path length (0.56 cm).
Statistical analysis
Statistical analyses were performed using SPSS 22.0 software. Data consisted of independent triplicates and were expressed as mean ± standard deviations. Statistical differences were assessed with Tukey’s test. The statistical significance was set at p < 0.05.
Results and discussion
Determination of enzymatic hydrolysis conditions
From the bioactive peptides with tyrosinase inhibition obtained from the hydrolysis of Pinctada martensii meat (PMHs) with proteases, B. licheniformis, neutral protease, flavourzyme, and compound proteinase were found to have the highest tyrosinase inhibition rates, with maximum rates of 21.5%, 14.5%, 18.5%, and 18.4%, respectively (Fig. 1). The hydrolysates obtained from the protease of B. licheniformis, neutral protease, pepsin, and compound proteinase had the highest peptide yields with 21.35%, 23.4%, 38%, and 20%, respectively. Thus, the protease from B. licheniformis was selected to hydrolyze P. martensii meat and generate tyrosinase inhibitory peptides. The influencing factors of enzymatic time, temperature, enzyme addition, pH, and material to water ratio, were optimized. As shown in Fig. 1, the peptide yield of PMHs increased to 20.43 ± 0.71%, and the 1.0 mg/mL of PMHs showed higher bioactivity, which makes the tyrosinase inhibition activity higher by 22.4 ± 0.32%, under the optimized enzymatic conditions (60 °C, 4 h, 3000 U/g of enzyme, pH = 8 and 1:4).

Effects of different proteases (A) and optimized hydrolysates conditions (B temperature; C time; D pH; E enzyme addition; F material water ratio) on the tyrosinase inhibition and peptide yield from P. martensii meat (p < 0.05)
Isolation and purification of tyrosinase inhibitory peptides
The biological activity of peptides could be significantly affected by their molecular weight. Hence, the use of ultrafiltration with different cutoff membranes to separate Pinctada martensii meat (PMHs), generated three fractions with different molecular weights: PMHs-1 (< 3 kDa), PMHs-2 (3–5 kDa), and PMHs-3 (> 5 kDa). As shown in Fig. 2A, comparing PMHs-1 to PMHs-2 (16.62%) and PMHs-3 (10.54%) suggested that PMHs-1 had a higher tyrosinase inhibitory capacity of 26.34%, consistent with previous studies that the peptides with smaller molecular weights exhibited higher biological activities (Atma et al. 2021; Cheng et al. 2021; Feng et al. 2021; Lin et al. 2017). Consequently, the five fractions obtained from PMHs-1 purification on the Sephadex G-15 column (Fig. 2B) suggested that the F4 fraction had the highest tyrosinase inhibitory activity (36.67 ± 1.13%) and was approximately 5.85- and 1.45 times more potent than the F3 and F5 fractions, respectively (Fig. 2C). Hence, the F4 fraction was collected and lyophilized for further identification.

Separation and purification of tyrosinase inhibitory peptides after the hydrolyzation of P. martensii meat: A peptides purified by ultrafiltration with different molecular weight cutoff; B gel filtration chromatography of PMHs-1 with Sephadex G-15 column; C determination of tyrosinase inhibitory capacity of each fraction (F1–F5). Peptide concentration was set as 1.0 mg/mL; all statistical tests (n = 3) were conducted, and values designated by different letters are considered statistically significant (p < 0.05)
Identification of amino acid sequences and molecular docking of tyrosinase inhibitory peptides
As shown in Fig. 3A, HPLC–MS analysis was conducted to determine the molecular weight and amino acid sequences of F4, wherein a total of 401 peptides were identified based on the base peak chromatogram. Among these, 28 peptides with confidence > 45 points were selected to dock with tyrosinase in the HPEPDOCK sever. The binding energies were also predicted by the PRODIGY webserver (Table 2), in which all peptides showed different binding energies to tyrosinase, ranging from −7.3 to −10.9 kcal/mol. These results were consistent with the results of previous studies where six peptides were identified from the chicken egg white proteins (Yap and Gan 2021). The three peptides with the lowest binding energy values, namely: WDRPKDDGGSPIK (W1, Twitchin-3 Partial BAN09026.1, −10.5 kcal/mol), DRGYPPVMF (W2, Hypothetical Protein JAS03782.1, −10.9 kcal/mol), and SGGGGGGGLGSGGSIRSSY (W3, −10.7 kcal/mol), were selected to be further analyzed.

Construction of molecular models between tyrosinase and peptides A 3D structure of the interactions between W1(A-1), W2(A-2), W3(A-3), and tyrosinase; B the 2D structure of the interactions of W1(B-1), W2(B-2), W3(B-3) with tyrosinase
Furthermore, the binding of W1–W3 to tyrosinase was analyzed using 2D and 3D diagrams to determine the binding sites and interaction forces between the peptides and tyrosinase. As shown in Fig. 2, W1–W3 exhibited varying binding degrees to the grooves of tyrosinase. Three kinds of interactions, hydrophobic interactions, hydrogen bonds, and salt bridges, were observed. Among these, several hydrogen bonds with Tyr79, His85, and Gly281 of tyrosinase and two salt bridges between the Arg3 residue and tyrosinase Glu322, and Lys5 residue and tyrosinase Glu189 were observed in W1. Meanwhile, W2 formed hydrogen bonds with Ala45, Q196, and T197 of tyrosinase, and W3 formed the highest number of hydrogen bonds with tyrosinase, including Gly223, Glu226, Pro270, Asn81, and Tyr65 (Table 2). W1 interacts with the active site of tyrosinase, while W2 and W3 do not bind to the active site. Although W2 and W3 did not bind to the active sites of tyrosinase, tyrosinase inhibitory activity may potentially occur (Deng et al. 2020; Yap and Gan 2021). Based on these results, the peptides of W1–W3 were selected to be further investigated as potential tyrosinase inhibitors.
In vitro activities of synthetic peptides
W1, W2, and W3 peptides were successfully synthesized according to their amino acid sequence. Thus, activities including tyrosinase inhibition, sun protection, and antioxidant activities were investigated in detail. A 5 mg/mL of sample concentration was used to assess the inhibitory activity of synthetic peptides. As shown in Table 3, the tyrosinase inhibition rate was consistent with the molecular docking, wherein W1 (12.86 ± 1.85%) and W2 (31.57 ± 0.97%) were far lower than that of W3 (60.07 ± 1.18%). This may be due to the higher number of hydrogen bonds between W3 and tyrosinase, as the salt bridges between Arg3 and Lys5, and Glu322 and Glu198 residues of tyrosinase were the lowest in W1. W3 had the most apparent tyrosinase inhibition activity as its IC50 was 3.04 mg/mL, which was higher than the pentapeptide NYRRE (IC50 = 9.77 mg/mL) of papaya seed protein (Deng et al. 2020) and the peptides from enzyme hydrolyzed royal jelly (IC50 = 7.59 mg/mL) (Ge et al. 2023). Therefore, W3 could be developed as a potential tyrosinase inhibitory peptide.
Due to the aromatic ring structure in the amino acid residues, tryptophan and tyrosine residues could cause strong UV absorption that results in higher SPF values (Cao and Zhao 2004; Stevanato et al. 2014). As shown in Table 3, three peptides had certain UV light absorption abilities. The SPF value of W1 was much higher than that of W2 and W3, with 5.47 ± 0.003, indicating that it is more suited to absorb UV radiation at a wavelength of 290–320 nm. However, the SPF value of W1 (11.9 ± 0.24 at 10 mg/mL) was still lower than that of the polypeptide purified from egg albumin (Yap and Gan 2021). Furthermore, due to the differences in the unique amino acid sequence and structure of peptides (Farajzadeh et al. 2021), the 5 mg/mL of W1, W2, and W3 showed different antioxidant capacities, wherein W1 had the most robust 2,2-diphenyl-1-picrylhydrazyl (DPPH)·scavenging capacity with 65.05 ± 0.256% (Table 3). In contrast, W3 exhibited the most potent 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic) acid (ABTS)· scavenging ability with 12.02 ± 0.633% and was the only peptide to display the ability to reduce Fe3+-tripyridyl triazine (Fe3+-TPTZ) to Fe2+-TPTZ (the total antioxidant capacity). Therefore, the peptides of W1, W2, and W3 could be exploited as multifunctional peptides.
Effects of synthetic peptides on B16-F10 cells
The effects of W2 and W3 on the survival rate, melanin content, and tyrosinase activity of mouse B16-F10 cells were investigated. As shown in Fig. 4, cell growth inhibition between W2 and W3 at different concentrations (1, 3, and 5 mg/mL) was not significantly different. For instance, when the concentration increased to 5 mg/mL after treatment, the cell survival rate remained at 90.22 ± 2.33% for W2 and 97.53 ± 1.32% for W3. This suggests that the concentration of W2 and W3 did not significantly affect the survival rate of the cells.

Effects of different components on B16-F10 cells: A cell viability, B intracellular melanin content, and C intracellular tyrosinase activity (p < 0.05)
Having tyrosinase inhibitory activity does not necessarily imply the ability to suppress melanin synthesis in mammalian cells. Previous studies have identified three active peptides with tyrosinase inhibitory effects, but in practice, only one of them has shown effective inhibition of melanin synthesis in mammalian cells (Ochiai et al. 2016). The different concentrations of W2 and W3 also effectively reduced the melanin content and tyrosinase activity in B16-F10 cells. The intracellular melanin content significantly decreased with increasing sample concentrations. Specifically, when the contents of W2 and W3 increased to 5 mg/mL, the relative contents of intracellular melanin also decreased from 100% to 81.62 ± 0.73% and 81.98 ± 1.34%, respectively, which was equivalent to that of arbutin at 1 mg/mL (the content of intracellular melanin was 81.16 ± 0.69%) (Fig. 4). Therefore, this demonstrates that the addition of W2 and W3 could effectively reduce the content of melanin in cells, or simply, they can induce whitening. Similarly, intracellular tyrosinase activity had significantly decreased with increasing peptide concentrations. The inhibitory effect of W2 on intracellular tyrosinase activity was much lower than that of arbutin (1.0 mg/mL, 19.19 ± 1.92%) but higher than W3 at the same concentration. When the concentrations of W2 and W3 increased to 5 mg/mL, the intracellular tyrosinase activity decreased to 30.70 ± 3.48% and 38.59 ± 0.96%, which was higher than the peptides from fish scale (59.73%) (Ju et al. 2022). This suggests that the intracellular tyrosinase activity can be more affected by W2 than W3.
Determination of the inhibition type and inhibition constant of W2 and W3
Reversibility of synthetic peptides inhibiting tyrosine monophenolase activity
The reversibility of synthetic peptides that inhibit tyrosine monophenolase activity was investigated by determining the effects of different polypeptide concentrations on the catalytic activity of tyrosine monophenolase through the continuous addition of substrate (l-tyrosine) and tyrosinase at different concentrations (Yu et al. 2021). The different concentrations of W2 and W3 resulted in a parallel linear relationship between the tyrosinase monophenolase concentration and enzyme activity, as shown in Fig. 5. The slope was also inversely proportional with the increase of W2 and W3 concentrations, indicating that the inhibition of W2 and W3 on tyrosinase monophenolase is reversible. Therefore, the effects of W2 and W3 on the catalytic efficiency of tyrosinase monophenolase could be achieved by inhibiting the free tyrosinase activity rather than reducing the content of effective enzymes in the system.

Reversibility analysis of tyrosinase by a W2 and b W3
Competitive types of synthetic peptides against tyrosinase and monophenolase
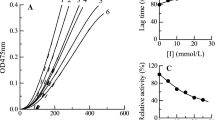
According to the previous study (Hridya et al. 2015), the Lineweaver Burk double reciprocal curve was drawn and shown in Fig. 6. With the increasing concentration of W2, four straight lines with different slopes intersected at the same point, and the intersection was located in the second quadrant, suggesting that W2 was a mixed inhibitor of tyrosine monophenolase that binds to both free enzyme and enzyme–substrate complex (Fig. 6A). The inhibition constant of the free enzyme (KI = 0.7576 mg/mL) and enzyme–substrate complex (KIS = 0.0351 mg/mL) was obtained by extrapolating the relationship curve between slope, intercept, and the corresponding W2 concentration. As a result, KI was much higher than KIS, indicating that W2 had a greater affinity for the free enzyme than enzyme–substrate complex (Yang et al. 2021).

Kinetic study of tyrosine monophenolase: A W2, n = 3; and B W3, n = 3
Similarly, when the concentration of W3 increased from 0 to 1.5 mg/mL, the slope of the straight line (Km/Vmax) gradually increased and intersected with the ordinate at the same point (1/Vmax), indicating that the change of W3 concentration did not affect Vmax (unchanged) (Fig. 6B). The inhibition constant KI was 2.48 mg /mL from the secondary plotting of the straight-line slope and W3 concentration. However, the value of Km gradually increased with the increase of W3 concentration, suggesting that W3 was a competitive inhibitor of tyrosine monophenolase that binds to the free enzyme. Inhibitors of enzymes commonly exhibit competitive inhibition, which typically shows dose-dependence (Yap and Gan 2020). Thus, by adjusting the inhibitor concentration, the inhibition rate can be controlled to some extent. Therefore, competitive inhibitory peptides have a wider range of applications (Thaha et al. 2021).
Conclusion
In this study, the tyrosinase inhibitory peptides were obtained through the enzymatic hydrolysis of P. martensii meat, separated, and purified using ultrafiltration and Sephadex G-15 column. Our results demonstrated that the F4 fraction exhibited the highest tyrosinase inhibitory activity (36.67 ± 1.13%). Hence, this fraction was collected and lyophilized for further identification. Subsequently, through the HPLC–MS analysis and molecular weight considerations, the three peptides, namely: WDRPKDDGGSPIK (W1, −10.5 kcal/mol), DRGYPPVMF (W2, −10.9 kcal/mol), and SGGGGGGGLGSGGSIRSSY (W3, −10.7 kcal/mol) had the lowest binding energy values. Hence, these peptides were selected and subjected to in vitro synthesis. W3 did not affect the survival rate of mouse melanoma B16-F10 cells at 1–5 mg/mL concentration but had a certain antioxidant capacity and SPF value that can effectively decrease melanin content by inhibiting intracellular tyrosinase activity. Thus, our study provides a novel pathway for utilizing marine organisms to obtain tyrosinase inhibitory peptides.
References
Atma Y, Fitriani D, Mustopa AZ (2021) Radical-scavenging activity of fish gelatin hydrolysates from bone of Pangasius catfish (Pangasius sutchi) by microbial proteases hydrolysis. Biointerface Res Appl Chem 11(1):7903–7911. https://doi.org/10.33263/briac111.79037911
Burnett CL, Bergfeld WF, Belsito DV, Hill RA, Klaassen CD, Liebler DC, Marks JG, Shank RC, Slaga TJ, Snyder PW, Andersen FA (2010) Final report of the safety assessment of kojic acid as used in cosmetics. Int J Toxicol 29:244S-273S. https://doi.org/10.1177/1091581810385956
Cao SX, Zhao YF (2004) Application of molecular absorption spectrophotometric method to the determination of biologic macromotecular structures. Spectrosc Spectr Anal 24(10):1197–1201
Cheng S, Tu M, Liu H, An Y, Du M, Zhu B (2021) A novel heptapeptide derived from Crassostrea gigas shows anticoagulant activity by targeting for thrombin active domain. Food Chem 334:127507
Deng YJ, Huang LX, Zhang CH, Xie PJ, Cheng J, Wang X, Liu LJ (2020) Skin-care functions of peptides prepared from Chinese quince seed protein: sequences analysis, tyrosinase inhibition and molecular docking study. Ind Crops Prod 148:112331. https://doi.org/10.1016/j.indcrop.2020.112331
Dutra EA, da Costa e Oliveira DAG, Kedor-Hackmann ERM, Santoro MIRM (2004) Determination of sun protection factor (SPF) of sunscreens by ultraviolet spectrophotometry [Determinação do fator de proteção solar (FPS) de protetores solares por espectrofotometria no ultravioleta]. Rev Bras Ciênc Farm 40(3):381–385. https://doi.org/10.1590/s1516-93322004000300014
Farajzadeh N, Saglam O, Akin M, Saki N, Kocak MB (2021) Investigation of tyrosinase enzyme (from mushroom) inhibitory activities and antioxidant properties of new fluorine-containing phthalocyanines. Arch Pharm 354(4):e2000340. https://doi.org/10.1002/ardp.202000340
Feng YX, Wang ZC, Chen JX, Li HR, Wang YB, Ren DF, Lu J (2021) Separation, identification, and molecular docking of tyrosinase inhibitory peptides from the hydrolysates of defatted walnut (Juglans regia L.) meal. Food Chem 353:129471. https://doi.org/10.1016/j.foodchem.2021.129471
Ge Z, Liu J-C, Sun J-A, Mao X-Z (2023) Tyrosinase inhibitory peptides from enzyme hydrolyzed royal jelly: production, separation, identification and docking analysis. Foods 12(11):2240
Gou L, Lee J, Hao H, Park YD, Zhan Y, Lü Z (2017) The effect of oxaloacetic acid on tyrosinase activity and structure: integration of inhibition kinetics with docking simulation. Int J Biol Macromol 101:59–66
Han TY, Son IP, Jang WS, Chang HS, Choi BS (2011) Efficacy of hydrogel mask with 2% arbutin for melasma. Korean J Dermatol 49(3):210–216
Hao JM (2007) Enzymolysis research on preparation of angitensin coverting enzyme inhibitory peptides from Pinctada martensii protein. Food Sci 28(5):128–131
Hao L, Wang X, Cao Y, Xu J, Xue C (2022) A comprehensive review of oyster peptides: preparation, characterisation and bioactivities. Rev Aquac 14(1):120–138
Harnedy PA, FitzGerald RJ (2012) Bioactive peptides from marine processing waste and shellfish: a review. J Funct Foods 4(1):6–24
Heriniaina RM, Dong J, Kalavagunta PK, Wu HL, Yan DS, Shang J (2018) Effects of six compounds with different chemical structures on melanogenesis. Chin J Nat Med 16(10):766–773. https://doi.org/10.1016/s1875-5364(18)30116-x
Hridya H, Amrita A, Sankari M, Doss CGP, Gopalakrishnan M, Gopalakrishnan C, Siva R (2015) Inhibitory effect of brazilein on tyrosinase and melanin synthesis: kinetics and in silico approach. Int J Biol Macromol 81:228–234. https://doi.org/10.1016/j.ijbiomac.2015.07.064
Jeon HJ, Kim K, Kim YD, Lee SE (2019) Antimelanogenic activities of piperlongumine derived from Piper longum on murine B16F10 melanoma cells in vitro and zebrafish embryos in vivo: its molecular mode of depigmenting action. Appl Biol Chem 62(1):61. https://doi.org/10.1186/s13765-019-0468-7
Ju X, Cheng S, Li H, Xu X, Wang Z, Du M (2022) Tyrosinase inhibitory effects of the peptides from fish scale with the metal copper ions chelating ability. Food Chem 390:133146
Kim YJ, Uyama H (2005) Tyrosinase inhibitors from natural and synthetic sources: structure, inhibition mechanism and perspective for the future. Cell Mol Life Sci 62(15):1707–1723. https://doi.org/10.1007/s00018-005-5054-y
Kim DH, Kim SJ, Ullah S, Yun HY, Chun P, Moon HR (2017) Design, synthesis, and antimelanogenic effects of (2-substituted phenyl-1,3-dithiolan-4-yl)methanol derivatives. Drug Des Dev Ther 11:827–836. https://doi.org/10.2147/dddt.S131538
Kim J, Kang S, Kwon H, Moon H, Park MC (2019) Dual functional bioactive-peptide, AIMP1-derived peptide (AdP), for anti-aging. J Cosmet Dermatol 18(1):251–257. https://doi.org/10.1111/jocd.12671
Lamiable A, Thevenet P, Rey J, Vavrusa M, Derreumaux P, Tuffery P (2016) PEP-FOLD3: faster denovo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res 44(W1):W449–W454. https://doi.org/10.1093/nar/gkw329
Langeswaran K, Suganya N, Sangavi P (2019) Study on recognition of novel RACK1 protein inhibitors for small cell lung cancer outlined by pharmacophore based virtual screening and molecular docking. Biocatal Agric Biotechnol 21:101301. https://doi.org/10.1016/j.bcab.2019.101301
Lee SW, Kim JH, Song H, Seok JK, Hong SS, Boo YC (2019) Luteolin 7-sulfate attenuates melanin synthesis through inhibition of CREB- and MITF-mediated tyrosinase expression. Antioxidants 8(4):87. https://doi.org/10.3390/antiox8040087
Lin K, Zhang L-W, Han X, Cheng D-Y (2017) Novel angiotensin I-converting enzyme inhibitory peptides from protease hydrolysates of Qula casein: quantitative structure-activity relationship modeling and molecular docking study. J Funct Foods 32:266–277
Liu XY, Huang L, Bai YX, Liu XL, Li SB (2020) Extracting bio-zinc and taurine from Pinctada martensii meat. J Food Sci 85(4):1125–1131. https://doi.org/10.1111/1750-3841.15055
Ochiai A, Tanaka S, Tanaka T, Taniguchi M (2016) Rice bran protein as a potent source of antimelanogenic peptides with tyrosinase inhibitory activity. J Nat Prod 79(10):2545–2551
O’Donoghue JL (2006) Hydroquinone and its analogues in dermatology—a risk-benefit viewpoint. J Cosmet Dermatol 5(3):196–203
Oh Y, Ahn CB, Je JY (2020) Ark shell protein-derived bioactive peptides promote osteoblastic differentiation through upregulation of the canonical Wnt/beta-catenin signaling in human bone marrow-derived mesenchymal stem cells. J Food Biochem 44(10):e13440. https://doi.org/10.1111/jfbc.13440
Park YS, Lee JY, Suh JS, Jin YM, Yu Y, Kim HY, Park YJ, Chung CP, Jo I (2014) Selective osteogenesis by a synthetic mineral inducing peptide for the treatment of osteoporosis. Biomaterials 35(37):9747–9754. https://doi.org/10.1016/j.biomaterials.2014.08.007
Park SH, Yi YS, Kim MY, Cho JY (2019) Antioxidative and antimelanogenesis effect of momordica charantia methanol extract. Evid Based Complement Alternat Med 2019:5091534. https://doi.org/10.1155/2019/5091534
Pilevneli AD, Konuklugil B (2020) Marine derived tyrosinase inhibitors. Su Urunleri Dergisi 37(4):427–436. https://doi.org/10.12714/egejfas.37.4.15
Stevanato R, Bertelle M, Fabris S (2014) Photoprotective characteristics of natural antioxidant polyphenols. Regul Toxicol Pharmacol 69(1):71–77
Thaha A, Wang B-S, Chang Y-W, Hsia S-M, Huang T-C, Shiau C-Y, Hwang D-F, Chen T-Y (2021) Food-derived bioactive peptides with antioxidative capacity, xanthine oxidase and tyrosinase inhibitory activity. Processes 9(5):747
Vangone A, Bonvin A (2017) PRODIGY: a contact-based predictor of binding affinity in protein-protein complexes. Bio-Protoc 7(3):e2124. https://doi.org/10.21769/BioProtoc.2124
Wang J, Yan-Yan WU, Lai-Hao LI, Yang XQ, Xiao HU (2014) Antioxidant stability and functional properties of Pinctada martensii protein hydrolysates. Sci Technol Food Ind 2014(13):67–71
Xiaowei Z, Jiang G, Li L, Ning Y (2004) Research developments in the general utilization of oyster. Mar Sci 69(2):331–338. https://doi.org/10.1113/expphysiol.1984.sp002809
Yan YM, Tao HY, He JH, Huang SY (2020) The HDOCK server for integrated protein-protein docking. Nat Protoc 15(5):1829–1852. https://doi.org/10.1038/s41596-020-0312-x
Yang D, Wang L, Zhai JX, Han N, Liu ZH, Li SK, Yin J (2021) Characterization of antioxidant, alpha-glucosidase and tyrosinase inhibitors from the rhizomes of Potentilla anserina L. and their structure-activity relationship. Food Chem 336:127714. https://doi.org/10.1016/j.foodchem.2020.127714
Yang F, Zhao D, Zhang K, Wang Z, Wang Y, Wu C, Cui S, Guo T, Chen L, Chen J (2022) Oral delivery of marine shellfish supramolecule peptides for skin wound healing. Colloids Surf B 216:112592
Yap P-G, Gan C-Y (2020) Chicken egg white-advancing from food to skin health therapy: optimization of hydrolysis condition and identification of tyrosinase inhibitor peptides. Foods 9(9):1312. https://doi.org/10.3390/foods9091312
Yap P-G, Gan C-Y (2021) Multifunctional tyrosinase inhibitor peptides with copper chelating, UV-absorption and antioxidant activities: kinetic and docking studies. Foods 10(3):675. https://doi.org/10.3390/foods10030675
Yu Q, Fan L (2021) Understanding the combined effect and inhibition mechanism of 4-hydroxycinnamic acid and ferulic acid as tyrosinase inhibitors. Food Chem 352(2):129369
Yu SY, He M, Zhai YH, Xie ZK, Xu S, Yu SX, Xiao HF, Song YD (2021) Inhibitory activity and mechanism of trilobatin on tyrosinase: kinetics, interaction mechanism and molecular docking. Food Funct 12(6):2569–2579. https://doi.org/10.1039/d0fo03264f
Zou K, Zhang D, Guo H, Zhang N, Jiang S (2015) Comparison of seven metallic elements in prismatic layer and nacreous layer of shells from four shell color strains in pearl oyster (Pinctada fucata). South China Fish Sci 11(3):74–79
Acknowledgements
This work was financially supported by the Key projects in Guangxi (Grant Number: 2019GXNSFDA245008), Development and Industrialization of Marine Health Nutrition Products (Grant Number: GUIKEAA17204075), Molecular Basis and Regulation Mechanism of Flavor Quality Formation in Aquation Products Processing (Grant Number: 20181119), and the “Bagui Scholars” Special Project.
Author information
Authors and Affiliations
Contributions
JM: literature searches and review, manuscript writing, figure design; JL and JL: literature searches and review, drawing; PJ: literature searches and review; XL: Supervision, Writing-review & editing; YB: Project administration; SL: literature searches and review, manuscript writing and revision.
Corresponding author
Ethics declarations
Conflict of interest
We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with this manuscript. We have no financial and personal relationships with other people or organizations that can inappropriately influence our work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Meng, J., Liu, J., Lu, J. et al. Isolation, identification, and preparation of tyrosinase inhibitory peptides from Pinctada martensii meat. Biotechnol Lett 45, 1495–1511 (2023). https://doi.org/10.1007/s10529-023-03437-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-023-03437-0